Information Theory of DNA Shotgun Sequencing Abolfazl S
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bioinformatics 1
Bioinformatics 1 Bioinformatics School School of Science, Engineering and Technology (http://www.stmarytx.edu/set/) School Dean Ian P. Martines, Ph.D. ([email protected]) Department Biological Science (https://www.stmarytx.edu/academics/set/undergraduate/biological-sciences/) Bioinformatics is an interdisciplinary and growing field in science for solving biological, biomedical and biochemical problems with the help of computer science, mathematics and information technology. Bioinformaticians are in high demand not only in research, but also in academia because few people have the education and skills to fill available positions. The Bioinformatics program at St. Mary’s University prepares students for graduate school, medical school or entry into the field. Bioinformatics is highly applicable to all branches of life sciences and also to fields like personalized medicine and pharmacogenomics — the study of how genes affect a person’s response to drugs. The Bachelor of Science in Bioinformatics offers three tracks that students can choose. • Bachelor of Science in Bioinformatics with a minor in Biology: 120 credit hours • Bachelor of Science in Bioinformatics with a minor in Computer Science: 120 credit hours • Bachelor of Science in Bioinformatics with a minor in Applied Mathematics: 120 credit hours Students will take 23 credit hours of core Bioinformatics classes, which included three credit hours of internship or research and three credit hours of a Bioinformatics Capstone course. BS Bioinformatics Tracks • Bachelor of Science -

13 Genomics and Bioinformatics
Enderle / Introduction to Biomedical Engineering 2nd ed. Final Proof 5.2.2005 11:58am page 799 13 GENOMICS AND BIOINFORMATICS Spencer Muse, PhD Chapter Contents 13.1 Introduction 13.1.1 The Central Dogma: DNA to RNA to Protein 13.2 Core Laboratory Technologies 13.2.1 Gene Sequencing 13.2.2 Whole Genome Sequencing 13.2.3 Gene Expression 13.2.4 Polymorphisms 13.3 Core Bioinformatics Technologies 13.3.1 Genomics Databases 13.3.2 Sequence Alignment 13.3.3 Database Searching 13.3.4 Hidden Markov Models 13.3.5 Gene Prediction 13.3.6 Functional Annotation 13.3.7 Identifying Differentially Expressed Genes 13.3.8 Clustering Genes with Shared Expression Patterns 13.4 Conclusion Exercises Suggested Reading At the conclusion of this chapter, the reader will be able to: & Discuss the basic principles of molecular biology regarding genome science. & Describe the major types of data involved in genome projects, including technologies for collecting them. 799 Enderle / Introduction to Biomedical Engineering 2nd ed. Final Proof 5.2.2005 11:58am page 800 800 CHAPTER 13 GENOMICS AND BIOINFORMATICS & Describe practical applications and uses of genomic data. & Understand the major topics in the field of bioinformatics and DNA sequence analysis. & Use key bioinformatics databases and web resources. 13.1 INTRODUCTION In April 2003, sequencing of all three billion nucleotides in the human genome was declared complete. This landmark of modern science brought with it high hopes for the understanding and treatment of human genetic disorders. There is plenty of evidence to suggest that the hopes will become reality—1631 human genetic diseases are now associated with known DNA sequences, compared to the less than 100 that were known at the initiation of the Human Genome Project (HGP) in 1990. -

A Clone-Array Pooled Shotgun Strategy for Sequencing Large Genomes
Downloaded from genome.cshlp.org on September 24, 2021 - Published by Cold Spring Harbor Laboratory Press Perspective A Clone-Array Pooled Shotgun Strategy for Sequencing Large Genomes Wei-Wen Cai,1,2 Rui Chen,1,2 Richard A. Gibbs,1,2,5 and Allan Bradley1,3,4 1Department of Molecular and Human Genetics, 2Human Genome Sequencing Center, and 3Howard Hughes Medical Institute, Baylor College of Medicine, Houston, Texas 77030, USA A simplified strategy for sequencing large genomes is proposed. Clone-Array Pooled Shotgun Sequencing (CAPSS) is based on pooling rows and columns of arrayed genomic clones,for shotgun library construction. Random sequences are accumulated,and the data are processed by sequential comparison of rows and columns to assemble the sequence of clones at points of intersection. Compared with either a clone-by-clone approach or whole-genome shotgun sequencing,CAPSS requires relatively few library constructions and only minimal computational power for a complete genome assembly. The strategy is suitable for sequencing large genomes for which there are no sequence-ready maps,but for which relatively high resolution STS maps and highly redundant BAC libraries are available. It is immediately applicable to the sequencing of mouse,rat,zebrafish, and other important genomes,and can be managed in a cooperative fashion to take advantage of a distributed international DNA sequencing capacity. Advances in DNA sequencing technology in recent years have Drosophila genome, and the computational requirements to greatly increased the throughput and reduced the cost of ge- perform the necessary pairwise comparisons increase approxi- nome sequencing. Sequencing of a complex genome the size mately as a square of the size of the genome (see Appendix). -

Sequence Assembly Demystified
REVIEWS COMPUTATIONAL TOOLS Sequence assembly demystified Niranjan Nagarajan1 and Mihai Pop2 Abstract | Advances in sequencing technologies and increased access to sequencing services have led to renewed interest in sequence and genome assembly. Concurrently, new applications for sequencing have emerged, including gene expression analysis, discovery of genomic variants and metagenomics, and each of these has different needs and challenges in terms of assembly. We survey the theoretical foundations that underlie modern assembly and highlight the options and practical trade-offs that need to be considered, focusing on how individual features address the needs of specific applications. We also review key software and the interplay between experimental design and efficacy of assembly. Paired-end data Previously the province of large genome centres, DNA between DNA sequences is then used to ‘stitch’ together Data from a pair of reads sequencing is now a key component of research carried the individual fragments into larger contiguous sequenced from ends of out in many laboratories. A number of new applica- sequences (contigs), thereby recovering the informa- the same DNA fragment. The tions of sequencing technologies have been spurred tion lost during the sequencing process. The assembly genomic distance between the reads is approximately by rapidly decreasing costs, including the study of process is complicated by the fact that, in many cases, known and is used to constrain microbial communities (metagenomics), the discovery this underlying assumption is incorrect. For example, assembly solutions. See also of structural variants in genomes and the analysis of genomic repeats — segments of DNA repeated in an ‘mate-pair read’. gene structure and expression. -

Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India Meera Yadav University of Delhi, [email protected]
University of Nebraska - Lincoln DigitalCommons@University of Nebraska - Lincoln Library Philosophy and Practice (e-journal) Libraries at University of Nebraska-Lincoln 2015 Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India meera yadav University of Delhi, [email protected] Manlunching Tawmbing Saha Institute of Nuclear Physisc, [email protected] Follow this and additional works at: http://digitalcommons.unl.edu/libphilprac Part of the Library and Information Science Commons yadav, meera and Tawmbing, Manlunching, "Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India" (2015). Library Philosophy and Practice (e-journal). 1254. http://digitalcommons.unl.edu/libphilprac/1254 Use of Bioinformatics Resources and Tools by Users of Bioinformatics Centers in India Dr Meera, Manlunching Department of Library and Information Science, University of Delhi, India [email protected], [email protected] Abstract Information plays a vital role in Bioinformatics to achieve the existing Bioinformatics information technologies. Librarians have to identify the information needs, uses and problems faced to meet the needs and requirements of the Bioinformatics users. The paper analyses the response of 315 Bioinformatics users of 15 Bioinformatics centers in India. The papers analyze the data with respect to use of different Bioinformatics databases and tools used by scholars and scientists, areas of major research in Bioinformatics, Major research project, thrust areas of research and use of different resources by the user. The study identifies the various Bioinformatics services and resources used by the Bioinformatics researchers. Keywords: Informaion services, Users, Inforamtion needs, Bioinformatics resources 1. Introduction ‘Needs’ refer to lack of self-sufficiency and also represent gaps in the present knowledge of the users. -

Challenges in Bioinformatics
Yuri Quintana, PhD, delivered a webinar in AllerGen’s Webinars for Research Success series on February 27, 2018, discussing different approaches to biomedical informatics and innovations in big-data platforms for biomedical research. His main messages and a hyperlinked index to his presentation follow. WHAT IS BIOINFORMATICS? Bioinformatics is an interdisciplinary field that develops analytical methods and software tools for understanding clinical and biological data. It combines elements from many fields, including basic sciences, biology, computer science, mathematics and engineering, among others. WHY DO WE NEED BIOINFORMATICS? Chronic diseases are rapidly expanding all over the world, and associated healthcare costs are increasing at an astronomical rate. We need to develop personalized treatments tailored to the genetics of increasingly diverse patient populations, to clinical and family histories, and to environmental factors. This requires collecting vast amounts of data, integrating it, and making it accessible and usable. CHALLENGES IN BIOINFORMATICS Data collection, coordination and archiving: These technologies have evolved to the point Many sources of biomedical data—hospitals, that we can now analyze not only at the DNA research centres and universities—do not have level, but down to the level of proteins. DNA complete data for any particular disease, due in sequencers, DNA microarrays, and mass part to patient numbers, but also to the difficulties spectrometers are generating tremendous of data collection, inter-operability -
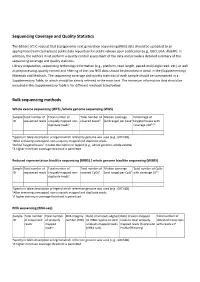
IJC Sequencing Coverage and Quality Statistics
Sequencing Coverage and Quality Statistics The Editors of IJC request that (epi)genomic next generation sequencing (NGS) data should be uploaded to an appropriate (restricted access) public data repository for public release upon publication (e.g., GEO, EGA, dbGAP). In addition, the authors must perform a quality control assessment of the data and provide a detailed summary of the sequencing coverage and quality statistics. Library preparation, sequencing technology information (e.g., platform, read length, paired‐end/single read, etc.) as well as preprocessing, quality control and filtering of the raw NGS data should be described in detail in the (Supplementary) Materials and Methods. The sequencing coverage and quality statistics of each sample should be summarized in a Supplementary Table, to which should be clearly referred in the main text. The minimum information that should be included in this Supplementary Table is for different methods listed below. Bulk sequencing methods Whole exome sequencing (WES) /whole genome sequencing (WGS) Sample Total number of Total number of Total number of Median coverage Percentage of ID sequenced reads uniquely mapped non‐ covered basesb (and range) per baseb targeted bases with duplicate readsa coverage ≥10b,c,d aSpecify in table description or legend which reference genome was used (e.g., GRCh38). bAfter removing unmapped, non‐uniquely mapped and duplicate reads. cDefine “targeted bases” in table description or legend (e.g., whole genome, whole exome). dA higher minimum coverage threshold is permitted. Reduced representation bisulfite sequencing (RRBS) / whole genome bisulfite sequencing (WGBS) Sample Total number of Total number of Total number of Median coverage Total number of CpGs ID sequenced reads uniquely mapped non‐ covered CpGsb (and range) per CpGb with coverage ≥5b,c duplicate readsa aSpecify in table description or legend which reference genome was used (e.g., GRCh38). -

Probabilities and Statistics in Shotgun Sequencing Shotgun Sequencing
Probabilities and Statistics in Shotgun Sequencing Shotgun Sequencing. Before any analysis of a DNA sequence can take place it is first necessary to determine the actual sequence itself, at least as accurately as is reasonably possible. Unfortunately, technical considerations make it impossible to sequence very long pieces of DNA all at once. Current sequencing technologies allow accurate reading of no more than 500 to 800bp of contiguous DNA sequence. This means that the sequence of an entire genome must be assembled from collections of comparatively short subsequences. This process is called DNA sequence “assembly ”. One approach of sequence assembly is to produce the sequence of a DNA segment (called as a “contig”, or perhaps a genome) from a large number of randomly chosen sequence reads (many overlapping small pieces, each on the order of 500-800 bases). One difficulty of this process is that the locations of the fragments within the genome and with respect to each other are not generally known. However, if enough fragments are sequenced so that there will be many overlaps between them, the fragments can be matched up and assembled. This method is called “ shotgun sequencing .” Shotgun sequencing approaches, including the whole-genome shotgun approach, are currently a central part of all genome-sequencing efforts. These methods require a high level of automation in sample preparation and analysis and are heavily reliant on the power of modern computers. There is an interplay between substrates to be sequenced (genomes and their representation in clone libraries), the analytical tools for generating a DNA sequence, the sequencing strategies, and the computational methods. -

Metagenomics Analysis of Microbiota by Next Generation Shotgun Sequencing
THE SWISS DNA COMPANY Application Note · Next Generation Sequencing Metagenomics Analysis of Microbiota by Next Generation Shotgun Sequencing Understand the genetic potential of your community samples Provides you with hypothesis-free taxonomic analysis Introduction Microbiome studies are often based on as the dependency on a single gene to microbiome analysis overcoming said the sequencing of specific marker genes analyze a whole community, the intro- limitations. Whole genomic DNA of as for instance the prokaryotic 16S rRNA duction of PCR bias and the restriction a sample is isolated, fragmented and gene. Such amplicon-based approaches to describe only the taxonomic compo- finally sequenced. This allows a detailed are well established and widely used. sition and diversity. Shotgun metage- analysis of the taxonomic and functional However, they have limitations such nomics is a cutting edge technique for composition of a microbial community. Microsynth Competences and Services Microsynth offers a full shotgun to your project requirements, thus pro- requirements to guarantee scientifically metagenomics service for taxonomic viding you with just the right amount of reliable results for your project. After and functional profiling of clinical, envi- data to answer your questions. quality processing the reads are aligned ronmental or engineered microbiomes. Bioinformatics: Taxonomic and func- against a protein reference database The service covers the entire process tional analysis of metagenomic data- (e.g. NCBI nr). Taxonomic and functional from experimental design, DNA isola- sets is challenging. Alignment, binning binning and annotation are performed. tion, tailored sequencing to detailed and annotation of the large amounts The analysis is not restricted to prokar- and customized bioinformatics analy- of sequencing reads require expertise yotes but also includes eukaryotes and sis. -

Comparing Bioinformatics Software Development by Computer Scientists and Biologists: an Exploratory Study
Comparing Bioinformatics Software Development by Computer Scientists and Biologists: An Exploratory Study Parmit K. Chilana Carole L. Palmer Amy J. Ko The Information School Graduate School of Library The Information School DUB Group and Information Science DUB Group University of Washington University of Illinois at University of Washington [email protected] Urbana-Champaign [email protected] [email protected] Abstract Although research in bioinformatics has soared in the last decade, much of the focus has been on high- We present the results of a study designed to better performance computing, such as optimizing algorithms understand information-seeking activities in and large-scale data storage techniques. Only recently bioinformatics software development by computer have studies on end-user programming [5, 8] and scientists and biologists. We collected data through semi- information activities in bioinformatics [1, 6] started to structured interviews with eight participants from four emerge. Despite these efforts, there still is a gap in our different bioinformatics labs in North America. The understanding of problems in bioinformatics software research focus within these labs ranged from development, how domain knowledge among MBB computational biology to applied molecular biology and and CS experts is exchanged, and how the software biochemistry. The findings indicate that colleagues play a development process in this domain can be improved. significant role in information seeking activities, but there In our exploratory study, we used an information is need for better methods of capturing and retaining use perspective to begin understanding issues in information from them during development. Also, in bioinformatics software development. We conducted terms of online information sources, there is need for in-depth interviews with 8 participants working in 4 more centralization, improved access and organization of bioinformatics labs in North America. -

DNA Sequencing
Contig Assembly ATCGATGCGTAGCAGACTACCGTTACGATGCCTT… TAGCTACGCATCGTCTGATGGCAATGCTACGGAA.. C T AG AGCAGA TAGCTACGCATCGT GT CTACCG GC TT AT CG GTTACGATGCCTT AT David Wishart, Ath 3-41 [email protected] DNA Sequencing 1 Principles of DNA Sequencing Primer DNA fragment Amp PBR322 Tet Ori Denature with Klenow + ddNTP heat to produce + dNTP + primers ssDNA The Secret to Sanger Sequencing 2 Principles of DNA Sequencing 5’ G C A T G C 3’ Template 5’ Primer dATP dATP dATP dATP dCTP dCTP dCTP dCTP dGTP dGTP dGTP dGTP dTTP dTTP dTTP dTTP ddCTP ddATP ddTTP ddGTP GddC GCddA GCAddT ddG GCATGddC GCATddG Principles of DNA Sequencing G T _ _ short C A G C A T G C + + long 3 Capillary Electrophoresis Separation by Electro-osmotic Flow Multiplexed CE with Fluorescent detection ABI 3700 96x700 bases 4 High Throughput DNA Sequencing Large Scale Sequencing • Goal is to determine the nucleic acid sequence of molecules ranging in size from a few hundred bp to >109 bp • The methodology requires an extensive computational analysis of raw data to yield the final sequence result 5 Shotgun Sequencing • High throughput sequencing method that employs automated sequencing of random DNA fragments • Automated DNA sequencing yields sequences of 500 to 1000 bp in length • To determine longer sequences you obtain fragmentary sequences and then join them together by overlapping • Overlapping is an alignment problem, but different from those we have discussed up to now Shotgun Sequencing Isolate ShearDNA Clone into Chromosome into Fragments Seq. Vectors Sequence 6 Shotgun Sequencing Sequence Send to Computer Assembled Chromatogram Sequence Analogy • You have 10 copies of a movie • The film has been cut into short pieces with about 240 frames per piece (10 seconds of film), at random • Reconstruct the film 7 Multi-alignment & Contig Assembly ATCGATGCGTAGCAGACTACCGTTACGATGCCTT… TAGCTACGCATCGTCTGATGGCAATGCTACGGAA. -

Database Technology for Bioinformatics from Information Retrieval to Knowledge Systems
Database Technology for Bioinformatics From Information Retrieval to Knowledge Systems Luis M. Rocha Complex Systems Modeling CCS3 - Modeling, Algorithms, and Informatics Los Alamos National Laboratory, MS B256 Los Alamos, NM 87545 [email protected] or [email protected] 1 Molecular Biology Databases 3 Bibliographic databases On-line journals and bibliographic citations – MEDLINE (1971, www.nlm.nih.gov) 3 Factual databases Repositories of Experimental data associated with published articles and that can be used for computerized analysis – Nucleic acid sequences: GenBank (1982, www.ncbi.nlm.nih.gov), EMBL (1982, www.ebi.ac.uk), DDBJ (1984, www.ddbj.nig.ac.jp) – Amino acid sequences: PIR (1968, www-nbrf.georgetown.edu), PRF (1979, www.prf.op.jp), SWISS-PROT (1986, www.expasy.ch) – 3D molecular structure: PDB (1971, www.rcsb.org), CSD (1965, www.ccdc.cam.ac.uk) Lack standardization of data contents 3 Knowledge Bases Intended for automatic inference rather than simple retrieval – Motif libraries: PROSITE (1988, www.expasy.ch/sprot/prosite.html) – Molecular Classifications: SCOP (1994, www.mrc-lmb.cam.ac.uk) – Biochemical Pathways: KEGG (1995, www.genome.ad.jp/kegg) Difference between knowledge and data (semiosis and syntax)?? 2 Growth of sequence and 3D Structure databases Number of Entries 3 Database Technology and Bioinformatics 3 Databases Computerized collection of data for Information Retrieval Shared by many users Stored records are organized with a predefined set of data items (attributes) Managed by a computer program: the database