Transcriptomics Technologies
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Bioconductor: Open Software Development for Computational Biology and Bioinformatics Robert C
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Collection Of Biostatistics Research Archive Bioconductor Project Bioconductor Project Working Papers Year 2004 Paper 1 Bioconductor: Open software development for computational biology and bioinformatics Robert C. Gentleman, Department of Biostatistical Sciences, Dana Farber Can- cer Institute Vincent J. Carey, Channing Laboratory, Brigham and Women’s Hospital Douglas J. Bates, Department of Statistics, University of Wisconsin, Madison Benjamin M. Bolstad, Division of Biostatistics, University of California, Berkeley Marcel Dettling, Seminar for Statistics, ETH, Zurich, CH Sandrine Dudoit, Division of Biostatistics, University of California, Berkeley Byron Ellis, Department of Statistics, Harvard University Laurent Gautier, Center for Biological Sequence Analysis, Technical University of Denmark, DK Yongchao Ge, Department of Biomathematical Sciences, Mount Sinai School of Medicine Jeff Gentry, Department of Biostatistical Sciences, Dana Farber Cancer Institute Kurt Hornik, Computational Statistics Group, Department of Statistics and Math- ematics, Wirtschaftsuniversitat¨ Wien, AT Torsten Hothorn, Institut fuer Medizininformatik, Biometrie und Epidemiologie, Friedrich-Alexander-Universitat Erlangen-Nurnberg, DE Wolfgang Huber, Department for Molecular Genome Analysis (B050), German Cancer Research Center, Heidelberg, DE Stefano Iacus, Department of Economics, University of Milan, IT Rafael Irizarry, Department of Biostatistics, Johns Hopkins University Friedrich Leisch, Institut fur¨ Statistik und Wahrscheinlichkeitstheorie, Technische Universitat¨ Wien, AT Cheng Li, Department of Biostatistical Sciences, Dana Farber Cancer Institute Martin Maechler, Seminar for Statistics, ETH, Zurich, CH Anthony J. Rossini, Department of Medical Education and Biomedical Informat- ics, University of Washington Guenther Sawitzki, Statistisches Labor, Institut fuer Angewandte Mathematik, DE Colin Smith, Department of Molecular Biology, The Scripps Research Institute, San Diego Gordon K. -

Statistical Computing with Pathway Tools Using Rcyc
Statistical Computing with Pathway Tools using RCyc Statistical Computing with Pathway Tools using RCyc Tomer Altman [email protected] Biomedical Informatics, Stanford University Statistical Computing with Pathway Tools using RCyc R & BioConductor S: software community over 30 years of statistical computing, data mining, machine learning, and data visualization knowledge R: open-source S with a lazy Scheme interpreter at its heart (including closures, symbols, and even macros!) RCyc: an R package to allow the interaction between Pathway / Genome Databases and the wealth of biostatistics software in the R community Statistical Computing with Pathway Tools using RCyc BioConductor Figure: BioConductor: Thousands of peer-reviewed biostatistics packages. Statistical Computing with Pathway Tools using RCyc Software`R'-chitecture C code extension to R to allow Unix socket access Common Lisp code to hack in XML-based communication Make the life of *Cyc API developers easier. Currently supports exchange of numbers, strings, and lists R code and documentation Provides utilities for starting PTools and marshaling data types Assumes user is familiar with the PTools API: http://bioinformatics.ai.sri.com/ptools/api/ All wrapped up in R package Easily installs via standard command-line R interface Statistical Computing with Pathway Tools using RCyc Simple Example callPToolsFn("so",list("'meta")) callPToolsFn("get-slot-value",list("'proton", "'common-name")) callPToolsFn("get-class-all-instances",list("'|Reactions|")) Statistical Computing with Pathway Tools using RCyc Availability http://github.com/taltman/RCyc Linked from PTools website Statistical Computing with Pathway Tools using RCyc Next Steps Dynamic instantiation of API functions in R Coming next release (coordination with BRG) Make development of *Cyc APIs easier, less boilerplate code Frame to Object import/export Provide \RCelot" functionality to slurp Ocelot frames directly into R S4 reference objects for direct data access Support for more exotic data types Symbols, hash tables, arrays, structures, etc. -

Proquesttm Pre-Made Cdna Libraries
Instruction Manual TM ProQuest Pre-made cDNA Libraries For detecting protein-protein interactions Version B December 12, 2002 25-0617 Table of Contents General Information ..............................................................2 Overview...............................................................................5 Using ProQuest™ Libraries....................................................8 pPC86.................................................................................13 pEXP-AD502.......................................................................15 Recipe.................................................................................17 Accessory Products ............................................................18 Purchaser Notification.........................................................19 Technical Service................................................................22 References..........................................................................24 1 General Information Contents 2 x 0.5 ml aliquots and Storage Each cDNA library is supplied in 80% SOB medium, 20% (v/v) glycerol. Store the library at -80°C. The cDNA library is stable for six months when stored properly. Titer of the Each library has greater than 5 x 109 cfu (colony Libraries forming units) derived from > 107 primary clones to ensure complete representation of rare sequences. ProQuest™ The following ProQuest™ Pre-made cDNA Libraries are Pre-made available from Invitrogen. For more information on cDNA preparing the library, see page -

In-Fusion Smarter Directional Cdna Library Construction Kit User Manual Table of Contents I
Takara Bio USA In-Fusion® SMARTer® Directional cDNA Library Construction Kit User Manual Cat. No. 634933 (050421) Takara Bio USA, Inc. 1290 Terra Bella Avenue, Mountain View, CA 94043, USA U.S. Technical Support: [email protected] United States/Canada Asia Pacific Europe Japan Page 1 of 40 800.662.2566 +1.650.919.7300 +33.(0)1.3904.6880 +81.(0)77.565.6999 In-Fusion SMARTer Directional cDNA Library Construction Kit User Manual Table of Contents I. List of Components .................................................................................................................................................. 4 II. Additional Materials Required .................................................................................................................................. 5 III. Introduction .......................................................................................................................................................... 6 IV. RNA Preparation & Handling ..............................................................................................................................11 A. Assessing the Quality of the RNA Template ........................................................................................................11 V. SMARTer cDNA Synthesis by LD-PCR .................................................................................................................13 A. Recommended Products ......................................................................................................................................13 -

Genome 559 Intro to Statistical and Computational Genomics
Genome 559! Intro to Statistical and Computational Genomics! Lecture 16a: Computational Gene Prediction Larry Ruzzo Today: Finding protein-coding genes coding sequence statistics prokaryotes mammals More on classes More practice Codons & The Genetic Code Ala : Alanine Second Base Arg : Arginine U C A G Asn : Asparagine Phe Ser Tyr Cys U Asp : Aspartic acid Phe Ser Tyr Cys C Cys : Cysteine U Leu Ser Stop Stop A Gln : Glutamine Leu Ser Stop Trp G Glu : Glutamic acid Leu Pro His Arg U Gly : Glycine Leu Pro His Arg C His : Histidine C Leu Pro Gln Arg A Ile : Isoleucine Leu Pro Gln Arg G Leu : Leucine Ile Thr Asn Ser U Lys : Lysine Ile Thr Asn Ser C Met : Methionine First Base A Third Base Ile Thr Lys Arg A Phe : Phenylalanine Met/Start Thr Lys Arg G Pro : Proline Val Ala Asp Gly U Ser : Serine Val Ala Asp Gly C Thr : Threonine G Val Ala Glu Gly A Trp : Tryptophane Val Ala Glu Gly G Tyr : Tyrosine Val : Valine Idea #1: Find Long ORF’s Reading frame: which of the 3 possible sequences of triples does the ribosome read? Open Reading Frame: No stop codons In random DNA average ORF = 64/3 = 21 triplets 300bp ORF once per 36kbp per strand But average protein ~ 1000bp So, coding DNA is not random–stops are rare Scanning for ORFs 1 2 3 U U A A U G U G U C A U U G A U U A A G! A A U U A C A C A G U A A C U A A U A C! 4 5 6 Idea #2: Codon Frequency,… Even between stops, coding DNA is not random In random DNA, Leu : Ala : Tryp = 6 : 4 : 1 But in real protein, ratios ~ 6.9 : 6.5 : 1 Even more: synonym usage is biased (in a species dependant way) Examples known with 90% AT 3rd base Why? E.g. -

Bioconductor: Open Software Development for Computational Biology and Bioinformatics Robert C
Bioconductor Project Bioconductor Project Working Papers Year 2004 Paper 1 Bioconductor: Open software development for computational biology and bioinformatics Robert C. Gentleman, Department of Biostatistical Sciences, Dana Farber Can- cer Institute Vincent J. Carey, Channing Laboratory, Brigham and Women’s Hospital Douglas J. Bates, Department of Statistics, University of Wisconsin, Madison Benjamin M. Bolstad, Division of Biostatistics, University of California, Berkeley Marcel Dettling, Seminar for Statistics, ETH, Zurich, CH Sandrine Dudoit, Division of Biostatistics, University of California, Berkeley Byron Ellis, Department of Statistics, Harvard University Laurent Gautier, Center for Biological Sequence Analysis, Technical University of Denmark, DK Yongchao Ge, Department of Biomathematical Sciences, Mount Sinai School of Medicine Jeff Gentry, Department of Biostatistical Sciences, Dana Farber Cancer Institute Kurt Hornik, Computational Statistics Group, Department of Statistics and Math- ematics, Wirtschaftsuniversitat¨ Wien, AT Torsten Hothorn, Institut fuer Medizininformatik, Biometrie und Epidemiologie, Friedrich-Alexander-Universitat Erlangen-Nurnberg, DE Wolfgang Huber, Department for Molecular Genome Analysis (B050), German Cancer Research Center, Heidelberg, DE Stefano Iacus, Department of Economics, University of Milan, IT Rafael Irizarry, Department of Biostatistics, Johns Hopkins University Friedrich Leisch, Institut fur¨ Statistik und Wahrscheinlichkeitstheorie, Technische Universitat¨ Wien, AT Cheng Li, Department -
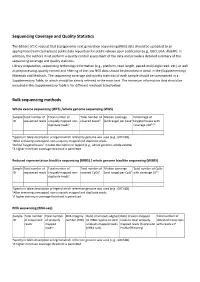
IJC Sequencing Coverage and Quality Statistics
Sequencing Coverage and Quality Statistics The Editors of IJC request that (epi)genomic next generation sequencing (NGS) data should be uploaded to an appropriate (restricted access) public data repository for public release upon publication (e.g., GEO, EGA, dbGAP). In addition, the authors must perform a quality control assessment of the data and provide a detailed summary of the sequencing coverage and quality statistics. Library preparation, sequencing technology information (e.g., platform, read length, paired‐end/single read, etc.) as well as preprocessing, quality control and filtering of the raw NGS data should be described in detail in the (Supplementary) Materials and Methods. The sequencing coverage and quality statistics of each sample should be summarized in a Supplementary Table, to which should be clearly referred in the main text. The minimum information that should be included in this Supplementary Table is for different methods listed below. Bulk sequencing methods Whole exome sequencing (WES) /whole genome sequencing (WGS) Sample Total number of Total number of Total number of Median coverage Percentage of ID sequenced reads uniquely mapped non‐ covered basesb (and range) per baseb targeted bases with duplicate readsa coverage ≥10b,c,d aSpecify in table description or legend which reference genome was used (e.g., GRCh38). bAfter removing unmapped, non‐uniquely mapped and duplicate reads. cDefine “targeted bases” in table description or legend (e.g., whole genome, whole exome). dA higher minimum coverage threshold is permitted. Reduced representation bisulfite sequencing (RRBS) / whole genome bisulfite sequencing (WGBS) Sample Total number of Total number of Total number of Median coverage Total number of CpGs ID sequenced reads uniquely mapped non‐ covered CpGsb (and range) per CpGb with coverage ≥5b,c duplicate readsa aSpecify in table description or legend which reference genome was used (e.g., GRCh38). -

Genbank Dennis A
D48–D53 Nucleic Acids Research, 2012, Vol. 40, Database issue Published online 5 December 2011 doi:10.1093/nar/gkr1202 GenBank Dennis A. Benson, Ilene Karsch-Mizrachi, Karen Clark, David J. Lipman, James Ostell and Eric W. Sayers* National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Building 38A, 8600 Rockville Pike, Bethesda, MD 20894, USA Received September 30, 2011; Revised November 14, 2011; Accepted November 17, 2011 ABSTRACT sequence (GSS), whole-genome shotgun (WGS) and Õ other high-throughput data from sequencing centers. GenBank is a comprehensive database that The US Office of Patents and Trademarks also contributes contains publicly available nucleotide sequences sequences from issued patents. GenBank participates with for more than 250 000 formally described species. the EMBL Nucleotide Sequence Database (EMBL-Bank), Downloaded from These sequences are obtained primarily through part of the European Nucleotide Archive (ENA) (2), and submissions from individual laboratories and batch the DNA Data Bank of Japan (DDBJ) (3) as a partner submissions from large-scale sequencing projects, in the International Nucleotide Sequence Database including whole-genome shotgun (WGS) and envir- Collaboration (INSDC). The INSDC partners exchange onmental sampling projects. Most submissions data daily to ensure that a uniform and comprehensive http://nar.oxfordjournals.org/ are made using the web-based BankIt or standalone collection of sequence information is available worldwide. Sequin programs, and accession numbers are NCBI makes the GenBank data available at no cost over the Internet, through FTP and a wide range of web-based assigned by GenBank staff upon receipt. Daily data retrieval and analysis services (4). -

Whole Genome Sequencing Data of Multiple Individuals of Pakistani
www.nature.com/scientificdata oPeN Whole genome sequencing data DAtA DeScriptor of multiple individuals of Pakistani descent Shahid Y. Khan1, Muhammad Ali1, Mei-Chong W. Lee2, Zhiwei Ma3, Pooja Biswas4, Asma A. Khan5, Muhammad Asif Naeem5, Saima Riazuddin 6, Sheikh Riazuddin5,7,8, Radha Ayyagari4, J. Fielding Hejtmancik 3 & S. Amer Riazuddin1 ✉ Here we report whole genome sequencing of four individuals (H3, H4, H5, and H6) from a family of Pakistani descent. Whole genome sequencing yielded 1084.92, 894.73, 1068.62, and 1005.77 million mapped reads corresponding to 162.73, 134.21, 160.29, and 150.86 Gb sequence data and 52.49x, 43.29x, 51.70x, and 48.66x average coverage for H3, H4, H5, and H6, respectively. We identifed 3,529,659, 3,478,495, 3,407,895, and 3,426,862 variants in the genomes of H3, H4, H5, and H6, respectively, including 1,668,024 variants common in the four genomes. Further, we identifed 42,422, 39,824, 28,599, and 35,206 novel variants in the genomes of H3, H4, H5, and H6, respectively. A major fraction of the variants identifed in the four genomes reside within the intergenic regions of the genome. Single nucleotide polymorphism (SNP) genotype based comparative analysis with ethnic populations of 1000 Genomes database linked the ancestry of all four genomes with the South Asian populations, which was further supported by mitochondria based haplogroup analysis. In conclusion, we report whole genome sequencing of four individuals of Pakistani descent. Background & Summary The completion of Human Genome Project ignited several large scale efforts to characterize variations in the human genome, which led to a comprehensive catalog of the common variants including single-nucleotide polymorphisms (SNPs) and insertions/deletions (indels), across the entire human genome1,2. -

Ijphs Jan Feb 07.Pmd
www.ijpsonline.com Review Article Pharmacogenomics and Computational Genomics: An Appraisal P. K. TRIPATHI*, SHALINI TRIPATHI AND N. K. JAIN1 Rajarshi Rananjay Sinh College of Pharmacy, Amethi-227 405, 1Department of Pharmaceutical Sciences, Dr. H. S. Gour Vishwavidyalaya, Sagar-470 003, India. Pharmacogenomics deals with the interactions of individual genetic constitution with drug therapy. It is very likely that pharmacogenetic tests will make up a significant proportion of total molecular biology testing in future. Therefore, this article emphasizes the applications of pharmacogenomics, and computational genome analysis in drug therapy. Patients sometimes experience adverse drug reactions for both efficacy and safety of a given drug regimen and (ADR) leading to deterioration of their underlying this is the central topic of pharmacogenomics. Drug condition in clinical practice. Physiology of individual development and pretreatment genetic analysis of patients patient can vary, so the response of an individual to drug will be two major practical aspects in pharmacogenomics. therapy may also be highly variable. This is a major Highly specialized drug research laboratories will achieve clinical problem, since this inter-individual variability is the first goal, while the second goal has to be achieved until now only partly predictable. Besides these medical by clinical .com).laboratories 2,3. problems, cost-benefit calculations for a given pharmacological therapy will be affected significantly. Drug development: This is exemplified by a recent US study, which estimates There are two pharmacogenomic approaches. First, for that over 100,000 patients die every year from ADRs1. most therapies a correct diagnosis is mandatory for a satisfactory therapeutic result. This is more relevant There are many factors such as age, sex, nutritional.medknow because many diseases may be caused by different status, kidney and liver function, concomitant diseases and genetic defects or be significantly affected by the genetic medications, and the disease that affects drug responses. -

Library Construction and Screening
Library construction and screening • A gene library is a collection of different DNA sequences from an organism, • which has beenAlso called genomic libraries or gene banks. • cloned into a vector for ease of purification, storage and analysis. Uses of gene libraries • To obtain the sequences of genes for analysis, amplification, cloning, and expression. • Once the sequence is known probes, primers, etc. can be synthesized for further diagnostic work using, for example, hybridization reactions, blots and PCR. • Knowledge of a gene sequence also offers the possibility of gene therapy. • Also, gene expression can be used to synthesize a product in particular host cells, e.g. synthesis of human gene products in prokaryotic cells. two types of gene library depending upon the source of the DNA used. 1.genomic library. 2.cDNA library Types of GENE library: • genomic library contains DNA fragments representing the entire genome of an organism. • cDNA library contains only complementary DNA molecules synthesized from mRNA molecules in a cell. Genomic Library : • Made from nuclear DNA of an organism or species. • DNA is cut into clonable size pieces as randomly possible using restriction endonuclease • Genomic libraries contain whole genomic fragments including gene exons and introns, gene promoters, intragenic DNA,origins of replication, etc Construction of Genomic Libraries 1. Isolation of genomic DNA and vector. 2.Cleavage of Genomic DNA and vector by Restriction Endonucleases. 3.Ligation of fragmented DNA with the vector. 4.Transformation of -

Next-Gen Sequencing Identifies Non-Coding Variation Disrupting
OPEN Molecular Psychiatry (2018) 23, 1375–1384 www.nature.com/mp ORIGINAL ARTICLE Next-gen sequencing identifies non-coding variation disrupting miRNA-binding sites in neurological disorders P Devanna1, XS Chen2,JHo1,2, D Gajewski1, SD Smith3, A Gialluisi2,4, C Francks2,5, SE Fisher2,5, DF Newbury6,7 and SC Vernes1,5 Understanding the genetic factors underlying neurodevelopmental and neuropsychiatric disorders is a major challenge given their prevalence and potential severity for quality of life. While large-scale genomic screens have made major advances in this area, for many disorders the genetic underpinnings are complex and poorly understood. To date the field has focused predominantly on protein coding variation, but given the importance of tightly controlled gene expression for normal brain development and disorder, variation that affects non-coding regulatory regions of the genome is likely to play an important role in these phenotypes. Herein we show the importance of 3 prime untranslated region (3'UTR) non-coding regulatory variants across neurodevelopmental and neuropsychiatric disorders. We devised a pipeline for identifying and functionally validating putatively pathogenic variants from next generation sequencing (NGS) data. We applied this pipeline to a cohort of children with severe specific language impairment (SLI) and identified a functional, SLI-associated variant affecting gene regulation in cells and post-mortem human brain. This variant and the affected gene (ARHGEF39) represent new putative risk factors for SLI. Furthermore, we identified 3′UTR regulatory variants across autism, schizophrenia and bipolar disorder NGS cohorts demonstrating their impact on neurodevelopmental and neuropsychiatric disorders. Our findings show the importance of investigating non-coding regulatory variants when determining risk factors contributing to neurodevelopmental and neuropsychiatric disorders.