Real Space in Situ Bond Energies: Toward a Consistent
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
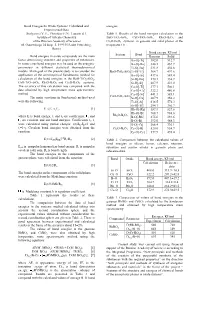
Bond Energies in Oxide Systems: Calculated and Experimental Data
Bond Energies In Oxide Systems: Calculated and energies. Experimental Data Stolyarova V. L., Plotnikov E.N., Lopatin S.I. Table 1. Results of the bond energies calculation in the Institute of Silicate Chemistry BaO-TiO 2-SiO 2, CaO-TiO 2-SiO 2, Rb 2O-B2O3 and of the Russian Academy of Sciences Cs 2O-B2O3 systems. in gaseous and solid phases at the ul. Odoevskogo 24 korp. 2, 199155 Sankt Petersburg, temperature 0 ¥ . Russia Bond energy, KJ/mol System Bond Bond energies in oxide compounds are the main Gaseous Solid factor determining structure and properties of substances. Ba-O[-Ti] 382.0 512.7 In some cases bond energies may be used as the energetic Ba-O[-Ba] 348.5 467.7 parameters in different statistical thermodynamical Ti-O[-Ba] 331.8 450.6 models. Main goal of the present study is to consider the BaO-TiO 2-SiO 2 Ti-O[-Ti] 364.7 497.6 application of the semiempirical Sanderson's method for Ba-O[-Si] 437.6 585.4 calculation of the bond energies in the BaO-TiO 2-SiO 2, Si-O[-Ba] 270.3 334.7 CaO-TiO 2-SiO 2, Rb 2O-B2O3 and Cs 2O-B2O3 systems. Si-O[-Si] 407.9 421.0 The accuracy of this calculation was compared with the Ca-O[-Ti] 377.1 500.1 data obtained by high temperature mass spectrometric Ca-O[-Ca] 322.2 486.8 method. Ca-O[-Si] 441.1 585.1 CaO-TiO -SiO The main equations in Sanderson's method used 2 2 Si-O[-Ca] 287.9 358.2 were the following. -

Molecular Orbital Theory to Predict Bond Order • to Apply Molecular Orbital Theory to the Diatomic Homonuclear Molecule from the Elements in the Second Period
Skills to Develop • To use molecular orbital theory to predict bond order • To apply Molecular Orbital Theory to the diatomic homonuclear molecule from the elements in the second period. None of the approaches we have described so far can adequately explain why some compounds are colored and others are not, why some substances with unpaired electrons are stable, and why others are effective semiconductors. These approaches also cannot describe the nature of resonance. Such limitations led to the development of a new approach to bonding in which electrons are not viewed as being localized between the nuclei of bonded atoms but are instead delocalized throughout the entire molecule. Just as with the valence bond theory, the approach we are about to discuss is based on a quantum mechanical model. Previously, we described the electrons in isolated atoms as having certain spatial distributions, called orbitals, each with a particular orbital energy. Just as the positions and energies of electrons in atoms can be described in terms of atomic orbitals (AOs), the positions and energies of electrons in molecules can be described in terms of molecular orbitals (MOs) A particular spatial distribution of electrons in a molecule that is associated with a particular orbital energy.—a spatial distribution of electrons in a molecule that is associated with a particular orbital energy. As the name suggests, molecular orbitals are not localized on a single atom but extend over the entire molecule. Consequently, the molecular orbital approach, called molecular orbital theory is a delocalized approach to bonding. Although the molecular orbital theory is computationally demanding, the principles on which it is based are similar to those we used to determine electron configurations for atoms. -

Calculating Lattice Energies Using the Born-Haber Cycle an Extension Activity for AP Chemistry Students
Calculating Lattice Energies Using the Born-Haber Cycle An Extension Activity for AP Chemistry Students A particular set of equations known as a Born-Haber cycle demonstrates how chemists are able to use the first law of thermodynamics—that the energy of the universe is conserved in any chemical or physical change—to find an unknown energy value that is difficult or impossible to measure experimentally. Some energy quantities, such as the lattice energy of a mineral or the electron affinity of an atom, can be difficult to measure in the lab. Examining a Born-Haber cycle we see that there is more than one path to the formation of a substance in a particular state, and that if we use consistent definitions, an energy value that we seek can be calculated from energy values that we already know. The following exercise will help us see the way these energy values relate to one another, give us practice with their definitions and symbols, and deepen our insight to their meaning when we see them in other types of problems. Each physical or chemical change represented has: • an equation that represents a clearly defined physical or chemical change; • a definition of the particular type of energy change; • a symbol or abbreviation for the energy change equal to a value for the change in enthalpy (ΔH), the energy that is released or absorbed during the change, expressed in kJ/mol; and • a name by which that change in enthalpy is commonly known. To set up the equations in a Born-Haber cycle, cut out the cards for names, equations, defini- tions, and symbols with energy values. -

The Theoretical Study on Interaction of Hydrogen with Single-Walled Boron Nitride Nanotubes
THE JOURNAL OF CHEMICAL PHYSICS 123, 114703 ͑2005͒ The theoretical study on interaction of hydrogen with single-walled boron nitride nanotubes. I. The reactive force field ReaxFFHBN development ͒ Sang Soo Han, Jeung Ku Kang, and Hyuck Mo Leea Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea Adri C. T. van Duin and William A. Goddard III Materials and Process Simulation Center, California Institute of Technology, California 91125 ͑Received 2 November 2004; accepted 21 June 2005; published online 19 September 2005͒ We present a new reactive force field ReaxFFHBN derived to accurately model large molecular and condensed phase systems of H, B, and N atoms. ReaxFFHBN has been tested against quantum calculation data for B–H, B–B, and B–N bond dissociations and for H–B–H, B–N–B, and N–B–N bond angle strain energies of various molecular clusters. The accuracy of the developed ReaxFFHBN for B–N–H systems is also tested for ͑i͒ H–B and H–B bond energies as a function of out of plane ͑ ͒ ͑ ͒ ͑ ͒ in H–B NH2 3 and H–N BH2 3, respectively, ii the reaction energy for the B3N3H6 +H2 → ͑ ͒ B3N3H8, and iii crystal properties such as lattice parameters and equations of states for the hexagonal type ͑h-BN͒ with a graphite structure and for the cubic type ͑c-BN͒ with a zinc-blende structure. For all these systems, ReaxFFHBN gives reliable results consistent with those from quantum calculations as it describes well bond breaking and formation in chemical processes and physical properties. -

Session #9: Homework Solutions
Session #9: Homework Solutions Problem #1 Estimate the ionic radius of Cs+. The lattice energy of CsCl is 633 kJ/mol. For CsCl the Madelung constant, M, is 1.763, and the Born exponent, n, is 10.7. The ionic radius of Cl– is known to be 1.81 Å. Solution Mq q ⎛⎞1 E1 = 1 2 −= and r r+r lattice ⎜⎟ o Cs+ Cl− 4rπεoo⎝⎠ n Solve first for ro MqqN ⎛⎞1 1.763(1.6 x 10-19 ) 2 6.02 x 1023 ⎛ 1 ⎞ r1 = 12 Aν −− = 1 o ⎜⎟ -12 5 ⎜ ⎟ 4Eπεolattice⎝⎠ n 4π 8.85 x 10 6.33x10 ⎝10.7⎠ = 3.50 x 10-10 m = 3.50 Å = r+ r Cs+- Cl ∴ r = 3.50-1.81 = 1.69 Å Cs+ Problem #2 (a) CFCs have been implicated in ozone depletion. Show that when Freon 12 (CCl2F2) is exposed to ultraviolet radiation, the compound decomposes to produce chlorine. (b) Draw the Lewis structure of Freon 12 and indicate the polarities of each bond within this compound. (c) Determine the percent ionic character of the C--Cl and C--F bonds. DATA: Average Bond Energies (kJ/mol) Single Bonds Multiple Bonds H-H 435 C = C 610 F-F 155 C ≡ C 836 Cl-Cl 242 C-C 347 Solution (a) Chlorine will be liberated if the C-Cl bond breaks, therefore, compute its strength and show that ultraviolet photons have enough energy to break the bond. 2 ECCl−−− =+ E CCx9 E ClCl 6.3(χχC− Cl) = 347 x 242 + 96.3(2.55 - 3.16)2 = 326 x 103/6.02 x 1023 = 5.41 x 10-19 J/bond photon will break ∴this bond if Eph > Ebond ⇒ critical λ is hc 6.6 x 10-34 x 3 x 108 λ = = = 3.66 x 10-7 m -19 Ebond 5.41 x 10 which lies in the u.v. -

The Nature of the Chemical Bond. Iv. the Energy of Single Bonds and the Relative Electronegativity of Atoms
3570 [CONTRIBUTION FROM THE GATES CHEMICAL LABORATORY, CALIFORNIA INSTITUE OF TECHNOLOGY, NO. 326] THE NATURE OF THE CHEMICAL BOND. IV. THE ENERGY OF SINGLE BONDS AND THE RELATIVE ELECTRONEGATIVITY OF ATOMS BY LINUS PAULING Journal of the American Chemical Society Volume 54, p. 3570-3582 September 1932 RECEIVED MAY 18, 1932 PUBLISHED SEPTEMBER 5, 1932 Recent developments in the application of the quantum mechanics to problems of molecular structure1 have indicated that the properties of a bond between two atoms often are determined mainly by one single-electron orbital wave function for each atom, and are not strongly affected by the other atoms in the molecule; for when the wave function for a molecule containing single covalent bonds is set up with the use of single-electron orbital wave functions, it is found that each bond function tends to overlap to the maximum extent the other function involved in one bond, and to avoid overlapping all others, so that the resonance integrals other than those characteristics of the bonds are small. The empirical evidence of interatomic distances supports this view; it has been possible to formulate a set of covalent radii for use in purely covalent molecules (in which each atom forms covalent bonds to a number determined by its position in the periodic system) which accounts satisfactory for observed distance in molecules to which a single Lewis electronic structure can be assigned.2 It has also been found that changing the covalence of an atom, and hence the nature of the bond function, produces a change in the radius.3 Independence of the bonds in a molecule would require that the total energy of formation of the molecule from separate atoms be expressible as the sum of constant energy terms characteristic of the various bonds. -
![Bonding Between Atoms [4]](https://docslib.b-cdn.net/cover/8940/bonding-between-atoms-4-2398940.webp)
Bonding Between Atoms [4]
Bonding between Atoms [4] ¾ Free atom: equilibrium at attractive and repulsive electrostatic forces between nucleus and electrosphere. • nucleus: protons (+) and neutrons • electrosphere: electrons (-) • electrical charge: 1.60 x 10-19 C • mass of proton: 1.67 x 10-24 g • mass of electron: 9.11 x 10-28 g Carbon atom Z=6 M=12.01 g/mol R=70 pm 1> Bonding between Atoms ¾ Interatomic bonds: forces that hold atoms together which act like little springs, linking one atom to the next in the solid state. ¾ Interatomic Forces: ¾ attractive forces (Fa) ¾ repulsive forces (Fr) ¾ The Coulomb's law: http://matterandinteractions.org/index.html q : Charge of object 1 q1 ⋅q2 1 F = K ⋅ q : Charge of object 2 e 2 2 r r: Distance between the two objects 9 2 -2 Ke = 8.9875 x 10 N.m .C (Coulomb Constant) F: Force between the two objects. F > 0 implies a repulsive interaction, while F < 0 means an attractive interaction. 2> Bonding between Atoms ¾ Consider two isolated atoms: – When the atoms are at large inter-atomic separation distance, the atoms do not exert any force on each other. – When the distance is decreased, an attractive force FA starts to act pulling atoms closer. – FA increases as the atoms gets closer. – But as the atoms get closer a repulsive force FR begin to act. – The net force FN between the two atoms is given by: FN = FA + FR –At some inter-atomic distance ro, FR exactly equals FA and FN becomes Zero FN = 0 = FA + FR –ro is called the equilibrium inter-atomic separation distance at which atoms enter into bonding 3> Bonding between Atoms ¾ equilibrium spacing: A B F = − F = A r p R r q where A, B, p, q are constants; q > p • When FA + FR = 0, equilibrium exists. -

Chapter 5 Molecular Orbitals
Chapter 5 Molecular Orbitals Molecular orbital theory uses group theory to describe the bonding in molecules ; it comple- ments and extends the introductory bonding models in Chapter 3 . In molecular orbital theory the symmetry properties and relative energies of atomic orbitals determine how these orbitals interact to form molecular orbitals. The molecular orbitals are then occupied by the available electrons according to the same rules used for atomic orbitals as described in Sections 2.2.3 and 2.2.4 . The total energy of the electrons in the molecular orbitals is compared with the initial total energy of electrons in the atomic orbitals. If the total energy of the electrons in the molecular orbitals is less than in the atomic orbitals, the molecule is stable relative to the separate atoms; if not, the molecule is unstable and predicted not to form. We will first describe the bonding, or lack of it, in the first 10 homonuclear diatomic molecules ( H2 through Ne2 ) and then expand the discussion to heteronuclear diatomic molecules and molecules having more than two atoms. A less rigorous pictorial approach is adequate to describe bonding in many small mole- cules and can provide clues to more complete descriptions of bonding in larger ones. A more elaborate approach, based on symmetry and employing group theory, is essential to under- stand orbital interactions in more complex molecular structures. In this chapter, we describe the pictorial approach and develop the symmetry methodology required for complex cases. 5.1 Formation of Molecular Orbitals from Atomic Orbitals As with atomic orbitals, Schrödinger equations can be written for electrons in molecules. -

Bond Dissociation Energies in Simple Molecules
2 A 11 10 145^03 j of Standards 0 NATL ' Bldg. 'Vmmiii SnSKBSllM^^'Wr Admin. All 102145903 1970 NSRDS-NBS 31 S S NBS-PUB-C 1964 QcToO U573 V31:1970 C.1 Bond Dissociation Energies In Simple Molecules — — ————- —— U.S. DEPARTMENT OF COMMERCE NSRDS NATIONAL BUREAU OF STANDARDS NATIONAL BUREAU OF STANDARDS The National Bureau of Standards 1 was established by an act of Congress March 3, 1901. Today, in addition to serving as the Nation’s central measurement laboratory, the Bureau is a principal focal point in the Federal Government for assuring maximum application of the physical and engineering sciences to the advancement of technology in industry and commerce. To this end the Bureau conducts research and provides central national services in four broad program areas. These are: (1) basic measurements and standards, (2) materials measurements and standards, (3) technological measurements and standards, and (4) transfer of technology. The Bureau comprises the Institute for Basic Standards, the Institute for Materials Research, the Institute for Applied Technology, the Center for Radiation Research, the Center for Computer Sciences and Technology, and the Office for Information Programs. THE INSTITUTE FOR BASIC STANDARDS provides the central basis within the United States of a complete and consistent system of physical measurement; coordinates that system with measurement systems of other nations; and furnishes essential services leading to accurate and uniform physical measurements throughout the Nation’s scientific community, industry, and com- merce. The Institute consists of an Office of Measurement Services and the following technical divisions: Applied Mathematics—Electricity—Metrology—Mechanics—Heat—Atomic and Molec- ular Physics—Radio Physics -—Radio Engineering -—Time and Frequency —Astro- physics - —Cryogenics. -

110 Exam5 Mccorkle SP16 Key
DO NOT OPEN UNTIL INSTRUCTED TO DO SO CHEM 110 – Dr. McCorkle – Exam #5 KEY While you wait, please complete the following information: Name: _______________________________ Student ID: _______________________________ Turn off cellphones and stow them away. No headphones, mp3 players, hats, sunglasses, food, drinks, restroom breaks, graphing calculators, programmable calculators, or sharing calculators. Grade corrections for incorrectly marked or incompletely erased answers will not be made. Dr. McCorkle CHEM 110 – Exam #5 KEY Spring 2016 2 Dr. McCorkle CHEM 110 – Exam #5 KEY Spring 2016 Multiple Choice – Choose the answer that best completes the question. Use an 815-E Scantron to record your response. [2 points each] 1. Which of the following statements is TRUE? A) A covalent bond is formed through the transfer of electrons from one atom to another. B) A pair of electrons involved in a covalent bond is sometimes referred to as a “lone pair.” C) It is not possible for two atoms to share more than two electrons. D) Single bonds are shorter than double bonds. E) In a covalent bond, the shared electrons interact with the nuclei of both the bonding atoms, thus lowering their potential energy. 2. Place the following in order of decreasing magnitude of lattice energy (largest to smallest). K2O Rb2S Li2O A) Li2O > K2O > Rb2S B) Li2O > Rb2S > K2O C) Rb2S > K2O > Li2O D) Rb2S > Li2O > K2O E) K2O > Li2O > Rb2S 3. Identify the weakest bond. A) single covalent bond B) double covalent bond C) triple covalent bond D) All of the above bonds are the same length 4. A reaction is exothermic when A) weak bonds break and strong bonds form. -

Kinetic Energy and the Covalent Bond in H2
1 / VOL. 2, NO. 6 ISSN 1430-4171 THE CHEMICAL EDUCATOR http://journals.springer-ny.com/chedr © 1997 SPRINGER-VERLAG NEW YORK, INC. 10.1007/s00897970153a In the Classroom Kinetic Energy and the Covalent + Bond in H2 FRANK RIOUX Department of Chemistry St. John’s University College of St. Benedict St. Joseph, MN 56374 [email protected] modified version of Ruedenberg’s innovative analysis of the chemical bond in the hydrogen Using a molecule ion is presented that factors the bond energy into bonding and nonbonding reasonably Acontributions. This simplified approach clearly illustrates accurate wave Ruedenberg’s main thesis: chemical bond formation is driven by a decrease in electron kinetic energy. function, he described the formation of the Introduction In 1962, Ruedenberg [1] published a persuasive quantum chemical bond in mechanical analysis of the hydrogen molecule ion that challenged the prevailing opinion that covalent bonding is terms of both simply an electrostatic phenomenon, by demonstrating the kinetic and crucial importance of kinetic energy in chemical bond formation. In the ensuing years, Ruedenberg, in collaboration potential energy. with others, extended and refined his original study [2–4]. While this important work has received some attention in the pedagogical literature [5–7], and brief treatment in a few 2 / VOL. 2, NO. 6 ISSN 1430-4171 THE CHEMICAL EDUCATOR http://journals.springer-ny.com/chedr © 1997 SPRINGER-VERLAG NEW YORK, INC. S 1430-4171 (97)06153-2 advanced texts [7–11], chemistry textbooks currently in use throughout the undergraduate curriculum present exclusively the conventional view that the covalent bond can be understood solely in terms of electrostatic potential energy. -

Hydrogen Bonding in Water
Hydrogen Bonding in Water The hydrogen bond in water is a dynamic attraction between neighboring water molecules involving one hydrogen atom located between the two oxygen atoms. Hydrogen bonds Introduction Water hydrogen bonds Water hydrogen bond length Water hydrogen bond direction Hydrogen bond cooperativity Water hydrogen bond 'wires' Rearranging hydrogen bonds Bifurcated hydrogen bonds Information transfer Hydrogen bonds and solubility 'Water is life' Audrey Hepburn Introduction Hydrogen bonding forms in liquid water as the hydrogen atoms of one water molecule are attracted towards the oxygen atom of a neighboring water molecule. In a water molecule (H2O), the oxygen nucleus with +8 charges attracts electrons better than the hydrogen nucleus with its +1 charge. Hence, the oxygen atom is partially negatively charged and the hydrogen atom is partially positively charged. The hydrogen atoms are not only covalently attached to their oxygen atoms but also attracted towards other nearby oxygen atoms. This attraction is the basis of the 'hydrogen' bonds. The water hydrogen bond is a weak bond, never stronger than about a twentieth of the strength of the O-H covalent bond. It is strong enough, however, to be maintained during thermal fluctuations at, and below, ambient temperatures. a The attraction of the O-H bonding electrons towards the oxygen atom leaves a deficiency on the far side of the hydrogen atom relative to the oxygen atom. The result is that the attractive force between the O-H hydrogen and the O-atom of a nearby water molecule is strongest when the three atoms are in close to a straight line and when the O-atoms are closer than 0.3 nm.