Calculating Lattice Energies Using the Born-Haber Cycle an Extension Activity for AP Chemistry Students
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 11: Solutions: Properties and Behavior
Chapter 11: Solutions: Properties and Behavior Learning Objectives 11.1: Interactions between Ions • Be able to estimate changes in lattice energy from the potential energy form of Coulomb’s Law, Q Q Cation Anion , in which is lattice energy, is a proportionality constant, is the charge on E = k E k QCation r the cation, Q is the charge on the anion, and r is the distance between the centers of the ions. Anion 11.2: Energy Changes during Formation and Dissolution of Ionic Compounds • Understand the concept of lattice energy and how it is a quantitative measure of stability for any ionic solid. As lattice energy increases, the stability of the ionic solid increases. • Be able to estimate changes in lattice energy from the potential energy form of Coulomb’s Law, Q Q Cation Anion , in which is lattice energy, is a proportionality constant, is the charge on E = k E k QCation r the cation, Q is the charge on the anion, and r is the distance between the centers of the ions. Anion • Be able to determine lattice energies using a Born-Haber cycle, which is a Hess’s Law process using ionization energies, electron affinities, and enthalpies for other atomic and molecular properties. • Be able to describe a molecular view of the solution process using Hess’s Law: 1. Solvent → Solvent Particles where ∆Hsolvent separation is endothermic. 2. Solute → Solute Particles where ∆Hsolute separation is endothermic. 3. Solvent Particles + Solute Particles → Solution where ∆Hsolvation can be endo- or exothermic. 4. ∆Hsolution = ∆Hsolvent separation + ∆Hsolute separation + ∆Hsolvation This is a Hess’s Law thinking process, and ∆Hsolvation is called the ∆Hhydration when the solvent is water. -

Assembly of Multi-Flavored Two-Dimensional Colloidal Crystals† Cite This: Soft Matter, 2017, 13,5397 Nathan A
Soft Matter View Article Online PAPER View Journal | View Issue Assembly of multi-flavored two-dimensional colloidal crystals† Cite this: Soft Matter, 2017, 13,5397 Nathan A. Mahynski, *a Hasan Zerze,b Harold W. Hatch, a Vincent K. Shena and Jeetain Mittal *b We systematically investigate the assembly of binary multi-flavored colloidal mixtures in two dimensions. In these mixtures all pairwise interactions between species may be tuned independently. This introduces an additional degree of freedom over more traditional binary mixtures with fixed mixing rules, which is anticipated to open new avenues for directed self-assembly. At present, colloidal self-assembly into non-trivial lattices tends to require either high pressures for isotropically interacting particles, or the introduction of directionally anisotropic interactions. Here we demonstrate tunable assembly into a plethora of structures which requires neither of these conditions. We develop a minimal model that defines a three-dimensional phase space containing one dimension for each pairwise interaction, then employ various computational techniques to map out regions of this phase space in which the system self-assembles into these different morphologies. We then present a mean-field model that is capable of reproducing these results for size-symmetric mixtures, which reveals how to target different structures by tuning pairwise interactions, solution stoichiometry, or both. Concerning particle size asymmetry, we find Received 19th May 2017, that domains in this model’s phase space, corresponding to different morphologies, tend to undergo a Accepted 30th June 2017 continuous ‘‘rotation’’ whose magnitude is proportional to the size asymmetry. Such continuity enables DOI: 10.1039/c7sm01005b one to estimate the relative stability of different lattices for arbitrary size asymmetries. -

CHAPTER 20: Lattice Energy
CHAPTER 20: Lattice Energy 20.1 Introduction to Lattice Energy 20.2 Born-Haber Cycles 20.3 Ion Polarisation 20.4 Enthalpy Changes in Solutions Learning outcomes: (a) explain and use the term lattice energy (∆H negative, i.e. gaseous ions to solid lattice). (b) explain, in qualitative terms, the effect of ionic charge and of ionic radius on the numerical magnitude of a lattice energy. (c) apply Hess’ Law to construct simple energy cycles, and carry out calculations involving such cycles and relevant energy terms, with particular reference to: (i) the formation of a simple ionic solid and of its aqueous solution. (ii) Born-Haber cycles (including ionisation energy and electron affinity). (d) interpret and explain qualitatively the trend in the thermal stability of the nitrates and carbonates in terms of the charge density of the cation and the polarisability of the large anion. (e) interpret and explain qualitatively the variation in solubility of Group II sulfates in terms of relative magnitudes of the enthalpy change of hydration and the corresponding lattice energy. 20.1 Introduction to Lattice Energy What is lattice energy? 1) In a solid ionic crystal lattice, the ions are bonded by strong ionic bonds between them. These forces are only completely broken when the ions are in gaseous state. 2) Lattice energy(or lattice enthalpy) is the enthalpy change when one mole of solid ionic lattice is formed from its scattered gaseous ions. 3) Lattice energy is always negative. This is because energy is always released when bonds are formed. 4) Use sodium chloride, NaCl as an example. -
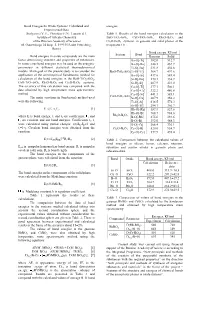
Bond Energies in Oxide Systems: Calculated and Experimental Data
Bond Energies In Oxide Systems: Calculated and energies. Experimental Data Stolyarova V. L., Plotnikov E.N., Lopatin S.I. Table 1. Results of the bond energies calculation in the Institute of Silicate Chemistry BaO-TiO 2-SiO 2, CaO-TiO 2-SiO 2, Rb 2O-B2O3 and of the Russian Academy of Sciences Cs 2O-B2O3 systems. in gaseous and solid phases at the ul. Odoevskogo 24 korp. 2, 199155 Sankt Petersburg, temperature 0 ¥ . Russia Bond energy, KJ/mol System Bond Bond energies in oxide compounds are the main Gaseous Solid factor determining structure and properties of substances. Ba-O[-Ti] 382.0 512.7 In some cases bond energies may be used as the energetic Ba-O[-Ba] 348.5 467.7 parameters in different statistical thermodynamical Ti-O[-Ba] 331.8 450.6 models. Main goal of the present study is to consider the BaO-TiO 2-SiO 2 Ti-O[-Ti] 364.7 497.6 application of the semiempirical Sanderson's method for Ba-O[-Si] 437.6 585.4 calculation of the bond energies in the BaO-TiO 2-SiO 2, Si-O[-Ba] 270.3 334.7 CaO-TiO 2-SiO 2, Rb 2O-B2O3 and Cs 2O-B2O3 systems. Si-O[-Si] 407.9 421.0 The accuracy of this calculation was compared with the Ca-O[-Ti] 377.1 500.1 data obtained by high temperature mass spectrometric Ca-O[-Ca] 322.2 486.8 method. Ca-O[-Si] 441.1 585.1 CaO-TiO -SiO The main equations in Sanderson's method used 2 2 Si-O[-Ca] 287.9 358.2 were the following. -

Molecular Orbital Theory to Predict Bond Order • to Apply Molecular Orbital Theory to the Diatomic Homonuclear Molecule from the Elements in the Second Period
Skills to Develop • To use molecular orbital theory to predict bond order • To apply Molecular Orbital Theory to the diatomic homonuclear molecule from the elements in the second period. None of the approaches we have described so far can adequately explain why some compounds are colored and others are not, why some substances with unpaired electrons are stable, and why others are effective semiconductors. These approaches also cannot describe the nature of resonance. Such limitations led to the development of a new approach to bonding in which electrons are not viewed as being localized between the nuclei of bonded atoms but are instead delocalized throughout the entire molecule. Just as with the valence bond theory, the approach we are about to discuss is based on a quantum mechanical model. Previously, we described the electrons in isolated atoms as having certain spatial distributions, called orbitals, each with a particular orbital energy. Just as the positions and energies of electrons in atoms can be described in terms of atomic orbitals (AOs), the positions and energies of electrons in molecules can be described in terms of molecular orbitals (MOs) A particular spatial distribution of electrons in a molecule that is associated with a particular orbital energy.—a spatial distribution of electrons in a molecule that is associated with a particular orbital energy. As the name suggests, molecular orbitals are not localized on a single atom but extend over the entire molecule. Consequently, the molecular orbital approach, called molecular orbital theory is a delocalized approach to bonding. Although the molecular orbital theory is computationally demanding, the principles on which it is based are similar to those we used to determine electron configurations for atoms. -

The Theoretical Study on Interaction of Hydrogen with Single-Walled Boron Nitride Nanotubes
THE JOURNAL OF CHEMICAL PHYSICS 123, 114703 ͑2005͒ The theoretical study on interaction of hydrogen with single-walled boron nitride nanotubes. I. The reactive force field ReaxFFHBN development ͒ Sang Soo Han, Jeung Ku Kang, and Hyuck Mo Leea Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea Adri C. T. van Duin and William A. Goddard III Materials and Process Simulation Center, California Institute of Technology, California 91125 ͑Received 2 November 2004; accepted 21 June 2005; published online 19 September 2005͒ We present a new reactive force field ReaxFFHBN derived to accurately model large molecular and condensed phase systems of H, B, and N atoms. ReaxFFHBN has been tested against quantum calculation data for B–H, B–B, and B–N bond dissociations and for H–B–H, B–N–B, and N–B–N bond angle strain energies of various molecular clusters. The accuracy of the developed ReaxFFHBN for B–N–H systems is also tested for ͑i͒ H–B and H–B bond energies as a function of out of plane ͑ ͒ ͑ ͒ ͑ ͒ in H–B NH2 3 and H–N BH2 3, respectively, ii the reaction energy for the B3N3H6 +H2 → ͑ ͒ B3N3H8, and iii crystal properties such as lattice parameters and equations of states for the hexagonal type ͑h-BN͒ with a graphite structure and for the cubic type ͑c-BN͒ with a zinc-blende structure. For all these systems, ReaxFFHBN gives reliable results consistent with those from quantum calculations as it describes well bond breaking and formation in chemical processes and physical properties. -

Session #9: Homework Solutions
Session #9: Homework Solutions Problem #1 Estimate the ionic radius of Cs+. The lattice energy of CsCl is 633 kJ/mol. For CsCl the Madelung constant, M, is 1.763, and the Born exponent, n, is 10.7. The ionic radius of Cl– is known to be 1.81 Å. Solution Mq q ⎛⎞1 E1 = 1 2 −= and r r+r lattice ⎜⎟ o Cs+ Cl− 4rπεoo⎝⎠ n Solve first for ro MqqN ⎛⎞1 1.763(1.6 x 10-19 ) 2 6.02 x 1023 ⎛ 1 ⎞ r1 = 12 Aν −− = 1 o ⎜⎟ -12 5 ⎜ ⎟ 4Eπεolattice⎝⎠ n 4π 8.85 x 10 6.33x10 ⎝10.7⎠ = 3.50 x 10-10 m = 3.50 Å = r+ r Cs+- Cl ∴ r = 3.50-1.81 = 1.69 Å Cs+ Problem #2 (a) CFCs have been implicated in ozone depletion. Show that when Freon 12 (CCl2F2) is exposed to ultraviolet radiation, the compound decomposes to produce chlorine. (b) Draw the Lewis structure of Freon 12 and indicate the polarities of each bond within this compound. (c) Determine the percent ionic character of the C--Cl and C--F bonds. DATA: Average Bond Energies (kJ/mol) Single Bonds Multiple Bonds H-H 435 C = C 610 F-F 155 C ≡ C 836 Cl-Cl 242 C-C 347 Solution (a) Chlorine will be liberated if the C-Cl bond breaks, therefore, compute its strength and show that ultraviolet photons have enough energy to break the bond. 2 ECCl−−− =+ E CCx9 E ClCl 6.3(χχC− Cl) = 347 x 242 + 96.3(2.55 - 3.16)2 = 326 x 103/6.02 x 1023 = 5.41 x 10-19 J/bond photon will break ∴this bond if Eph > Ebond ⇒ critical λ is hc 6.6 x 10-34 x 3 x 108 λ = = = 3.66 x 10-7 m -19 Ebond 5.41 x 10 which lies in the u.v. -

A.P. Chemistry Quiz: Ch. 8, Bonding Lattice Energy, Bond Enthalpy, Electronegativity, and Polarity Name___
A.P. Chemistry Quiz: Ch. 8, Bonding Lattice Energy, Bond Enthalpy, Electronegativity, and Polarity Name___________________________________ MULTIPLE CHOICE. Choose the one alternative that best completes the statement or answers the question. The diagram below is the Born-Haber cycle for the formation of crystalline potassium fluoride. 1) Which energy change corresponds to the negative lattice energy of potassium fluoride? A) 5 B) 1 C) 6 D) 4 E) 2 2) Which energy change corresponds to the electron affinity of fluorine? A) 2 B) 5 C) 6 D) 4 E) 1 3) Which energy change corresponds to the first ionization energy of potassium? A) 2 B) 4 C) 3 D) 5 E) 6 4) Given the electronegativities below, which covalent single bond is most polar? Element: H C N O Electronegativity: 2.1 2.5 3.0 3.5 A) C-H B) O-C C) O-H D) N-H E) O-N 5) Of the molecules below, the bond in __________ is the most polar. A) HBr B) HCl C) HF D) HI E) H2 A-1 6) Using the table of average bond energies below, the DH for the reaction is __________ kJ. Bond: C”C C-C H-I C-I C-H D (kJ/mol): 839 348 299 240 413 A) +160 B) -63 C) +63 D) -160 E) -217 7) Of the bonds C-N, ChN, and C”N, the C-N bond is __________. A) strongest/longest B) weakest/longest C) intermediate in both strength and length D) strongest/shortest E) weakest/shortest A-2 Answer Key Testname: QUIZ_BONDING_CH_08.TST MULTIPLE CHOICE. -

Real Space in Situ Bond Energies: Toward a Consistent
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repositorio Institucional de la Universidad de Oviedo Real space in situ bond energies: toward a consistent energetic definition of bond strength D. Menéndez-Crespo, A. Costales, E. Francisco, and A. Martín Pendása∗ June 20, 2018 Abstract A rigorous definition of intrinsic bond strength based on the partitioning of a molecule into real space fragments is presented. Using the domains provided by the quantum theory of atoms in molecules (QTAIM) together with the interacting quantum atoms (IQA) energetic decomposition, we show how an in situ bond strength, matching all the requirements of an intrinsic bond energy, can be defined between each pair of fragments. Total atomization or fragmentation energies are shown to be equal to the sum of these in situ bond energies (ISBEs) if the energies of the fragments are measured with respect to their in-the-molecule state. These energies usually lie above the ground state of the isolated fragments by quantities identified with the standard frag- ment relaxation or deformation energies, which are also provided by the protocol. Deformation energies bridge dissociation energies with ISBEs, and can be dissected using well-known tools of real space theories of chemical bonding. Similarly, ISBEs can be partitioned into ionic and covalent contributions, and this feature adds to the chemical appeal of the procedure. All the energetic quantities examined are observable and amenable, in principle, to experimental deter- mination. Several systems, exemplifying the role of each energetic term herein presented are used to show the power of the approach. -

Solutions Thermodynamics Dcians
Solutions Thermodynamics DCI Name _____________________________ Section ______ 1. The three attractive interactions which are important in solution formation are; solute-solute interactions, solvent-solvent interactions, and solute-solvent interactions. Define each of these interactions and describe their importance in determining whether a particular solute-solvent pair will form a homogeneous mixture or a heterogeneous mixture. Solute-solute interactions are the intermolecular attractions between solute particles. Solvent-solvent interactions are the intermolecular attractions between solvent particles. Solute-solvent interactions are the intermolecular attractions between a solute particle and a solvent particle. If the intermolecular attractions between solute particles are different compared to the intermolecular attractions between solvent particles it is unlikely dissolution will occur. Similar intermolecular attractions between solute particles and between solvent particles make it more likely a solution will form when the two are mixed. Shown below are two beakers. One contains liquid The two liquids form a water and the other contains liquid carbon heterogeneous mixture. The tetrachloride. For simplicity a circle is used to drawing should show the carbon represent the molecule. Complete the beaker on the tetrachloride on the bottom and right to illustrate the result of mixing the two liquids. the water on top, due to the difference in densities. Shown below are two beakers. One contains liquid The two liquids form a solution. hexane and the other contains liquid carbon The two components are tetrachloride. For simplicity a circle is used to uniformly distributed represent the molecule. Complete the beaker on the throughout the solution. right to illustrate the result of mixing the two liquids. -

Stoichiometric Control of DNA-Grafted Colloid Self-Assembly
Stoichiometric control of DNA-grafted colloid self-assembly Thi Voa, Venkat Venkatasubramaniana, Sanat Kumara,1, Babji Srinivasanb, Suchetan Pala,c, Yugang Zhangc, and Oleg Gangc aDepartment of Chemical Engineering, Columbia University, New York, NY 10027; bDepartment of Chemical Engineering, Indian Institute of Technology, Gandhinagar 382424, India; and cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, NY 11973 Edited by Monica Olvera de la Cruz, Northwestern University, Evanston, IL, and approved March 3, 2015 (received for review October 31, 2014) There has been considerable interest in understanding the self- reinforces the commonly held view that the desired crystal will only assembly of DNA-grafted nanoparticles into different crystal struc- form if the right stoichiometry is used. tures, e.g., CsCl, AlB2, and Cr3Si. Although there are important Similarly, several coarse-grained models also allow for the exceptions, a generally accepted view is that the right stoichiometry rapid determination of the crystal structure formed by binary of the two building block colloids needs to be mixed to form the mixtures of DNA-grafted colloids (23–25). The complementary desired crystal structure. To incisively probe this issue, we combine contact model (CCM) is a canonical example of such a coarse- experiments and theory on a series of DNA-grafted nanoparticles at grained description (23). Inspired by experiments, this theory varying stoichiometries, including noninteger values. We show that assumes that only attractive energetics (derived from DNA stoichiometry can couple with the geometries of the building blocks base pairing) determine the standard-state free energy. Thus, to tune the resulting equilibrium crystal morphology. As a concrete the CCM ignores any repulsive interactions. -

Arxiv:Cond-Mat/0602428V1
Ground-State Shapes and Structures of Colloidal Domains Jianlan Wu and Jianshu Cao∗ Department of Chemistry, Massachusetts Institute of Technology Cambridge, MA 02139 (Dated: First submitted August 21, 2004) Abstract In charged colloidal suspensions, the competition between square-well attraction and long-range Yukawa repulsion leads to various stable domains and Wigner supercrystals. Using a continuum model and symmetry arguments, a phase diagram of spheres, cylinders, and lamellae is obtained as a function of two control parameters, the volume fraction and the ratio between the surface tension and repulsion. Above a critical value of the ratio, the microphase cannot be supported by the Yukawa repulsion and macroscopic phase separation occurs. This finding quantitatively explains the lack of pattern formation in simple liquids because of the small hard sphere diameter in comparison with the size of macromolecules. The phase diagram also predicts microphase separation at zero value of the ratio, suggesting the possibility of self-assembly in repulsive systems. PACS numbers: 82.70.Dd, 05.65.+b, 64.70.-p arXiv:cond-mat/0602428v1 [cond-mat.soft] 17 Feb 2006 ∗ Email: [email protected] 1 Microphase separation is ubiquitous in soft matter systems[1, 2, 3, 4, 5, 6, 7, 8]. For example, microphase separation in block copolymers results from mixing of two or more chemically different polymer segments[3]. Competition of hydrophobic and hydrophilic head- groups of amphiphiles leads to micelles in the water solution[4]. The self-assembly processes in charged colloidal suspensions and protein solutions can lead to the formation of stable domains and Wigner supercrystals (see Fig.