Molecular Orbital Theory to Predict Bond Order • to Apply Molecular Orbital Theory to the Diatomic Homonuclear Molecule from the Elements in the Second Period
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Direct Preparation of Some Organolithium Compounds from Lithium and RX Compounds Katashi Oita Iowa State College
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1955 Direct preparation of some organolithium compounds from lithium and RX compounds Katashi Oita Iowa State College Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Oita, Katashi, "Direct preparation of some organolithium compounds from lithium and RX compounds " (1955). Retrospective Theses and Dissertations. 14262. https://lib.dr.iastate.edu/rtd/14262 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overiaps. -

The Global Potential Energy Surfaces of the Lowest Two 1A' States of The
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1992 The global potential energy surfaces of the lowest two 1A' states of the ozone molecule: theoretical determination and analysis Gregory John Atchity Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Atomic, Molecular and Optical Physics Commons, and the Physical Chemistry Commons Recommended Citation Atchity, Gregory John, "The global potential energy surfaces of the lowest two 1A' states of the ozone molecule: theoretical determination and analysis " (1992). Retrospective Theses and Dissertations. 10093. https://lib.dr.iastate.edu/rtd/10093 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly firom the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. -

Mineral Processing
Mineral Processing Foundations of theory and practice of minerallurgy 1st English edition JAN DRZYMALA, C. Eng., Ph.D., D.Sc. Member of the Polish Mineral Processing Society Wroclaw University of Technology 2007 Translation: J. Drzymala, A. Swatek Reviewer: A. Luszczkiewicz Published as supplied by the author ©Copyright by Jan Drzymala, Wroclaw 2007 Computer typesetting: Danuta Szyszka Cover design: Danuta Szyszka Cover photo: Sebastian Bożek Oficyna Wydawnicza Politechniki Wrocławskiej Wybrzeze Wyspianskiego 27 50-370 Wroclaw Any part of this publication can be used in any form by any means provided that the usage is acknowledged by the citation: Drzymala, J., Mineral Processing, Foundations of theory and practice of minerallurgy, Oficyna Wydawnicza PWr., 2007, www.ig.pwr.wroc.pl/minproc ISBN 978-83-7493-362-9 Contents Introduction ....................................................................................................................9 Part I Introduction to mineral processing .....................................................................13 1. From the Big Bang to mineral processing................................................................14 1.1. The formation of matter ...................................................................................14 1.2. Elementary particles.........................................................................................16 1.3. Molecules .........................................................................................................18 1.4. Solids................................................................................................................19 -
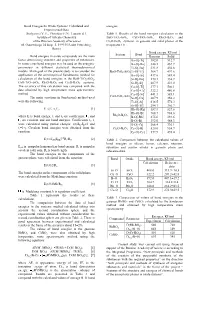
Bond Energies in Oxide Systems: Calculated and Experimental Data
Bond Energies In Oxide Systems: Calculated and energies. Experimental Data Stolyarova V. L., Plotnikov E.N., Lopatin S.I. Table 1. Results of the bond energies calculation in the Institute of Silicate Chemistry BaO-TiO 2-SiO 2, CaO-TiO 2-SiO 2, Rb 2O-B2O3 and of the Russian Academy of Sciences Cs 2O-B2O3 systems. in gaseous and solid phases at the ul. Odoevskogo 24 korp. 2, 199155 Sankt Petersburg, temperature 0 ¥ . Russia Bond energy, KJ/mol System Bond Bond energies in oxide compounds are the main Gaseous Solid factor determining structure and properties of substances. Ba-O[-Ti] 382.0 512.7 In some cases bond energies may be used as the energetic Ba-O[-Ba] 348.5 467.7 parameters in different statistical thermodynamical Ti-O[-Ba] 331.8 450.6 models. Main goal of the present study is to consider the BaO-TiO 2-SiO 2 Ti-O[-Ti] 364.7 497.6 application of the semiempirical Sanderson's method for Ba-O[-Si] 437.6 585.4 calculation of the bond energies in the BaO-TiO 2-SiO 2, Si-O[-Ba] 270.3 334.7 CaO-TiO 2-SiO 2, Rb 2O-B2O3 and Cs 2O-B2O3 systems. Si-O[-Si] 407.9 421.0 The accuracy of this calculation was compared with the Ca-O[-Ti] 377.1 500.1 data obtained by high temperature mass spectrometric Ca-O[-Ca] 322.2 486.8 method. Ca-O[-Si] 441.1 585.1 CaO-TiO -SiO The main equations in Sanderson's method used 2 2 Si-O[-Ca] 287.9 358.2 were the following. -

Introduction to Molecular Orbital Theory
Chapter 2: Molecular Structure and Bonding Bonding Theories 1. VSEPR Theory 2. Valence Bond theory (with hybridization) 3. Molecular Orbital Theory ( with molecualr orbitals) To date, we have looked at three different theories of molecular boning. They are the VSEPR Theory (with Lewis Dot Structures), the Valence Bond theory (with hybridization) and Molecular Orbital Theory. A good theory should predict physical and chemical properties of the molecule such as shape, bond energy, bond length, and bond angles.Because arguments based on atomic orbitals focus on the bonds formed between valence electrons on an atom, they are often said to involve a valence-bond theory. The valence-bond model can't adequately explain the fact that some molecules contains two equivalent bonds with a bond order between that of a single bond and a double bond. The best it can do is suggest that these molecules are mixtures, or hybrids, of the two Lewis structures that can be written for these molecules. This problem, and many others, can be overcome by using a more sophisticated model of bonding based on molecular orbitals. Molecular orbital theory is more powerful than valence-bond theory because the orbitals reflect the geometry of the molecule to which they are applied. But this power carries a significant cost in terms of the ease with which the model can be visualized. One model does not describe all the properties of molecular bonds. Each model desribes a set of properties better than the others. The final test for any theory is experimental data. Introduction to Molecular Orbital Theory The Molecular Orbital Theory does a good job of predicting elctronic spectra and paramagnetism, when VSEPR and the V-B Theories don't. -

Paul B. Tomascak Tomáš Magna Ralf Dohmen Advances in Lithium Isotope Geochemistry Advances in Isotope Geochemistry
Advances in Isotope Geochemistry Paul B. Tomascak Tomáš Magna Ralf Dohmen Advances in Lithium Isotope Geochemistry Advances in Isotope Geochemistry Series editor Jochen Hoefs, Göttingen, Germany More information about this series at http://www.springer.com/series/8152 Paul B. Tomascak • Tomáš Magna Ralf Dohmen Advances in Lithium Isotope Geochemistry 123 Paul B. Tomascak Ralf Dohmen State University of New York at Oswego Institute of Geology, Mineralogy Oswego, NY and Geophysics USA Ruhr-University Bochum Bochum, Nordrhein-Westfalen Tomáš Magna Germany Czech Geological Survey Prague Czech Republic ISSN 2364-5105 ISSN 2364-5113 (electronic) Advances in Isotope Geochemistry ISBN 978-3-319-01429-6 ISBN 978-3-319-01430-2 (eBook) DOI 10.1007/978-3-319-01430-2 Library of Congress Control Number: 2015953806 Springer Cham Heidelberg New York Dordrecht London © Springer International Publishing Switzerland 2016 This work is subject to copyright. All rights are reserved by the Publisher, whether the whole or part of the material is concerned, specifically the rights of translation, reprinting, reuse of illustrations, recitation, broadcasting, reproduction on microfilms or in any other physical way, and transmission or information storage and retrieval, electronic adaptation, computer software, or by similar or dissimilar methodology now known or hereafter developed. The use of general descriptive names, registered names, trademarks, service marks, etc. in this publication does not imply, even in the absence of a specific statement, that such names are exempt from the relevant protective laws and regulations and therefore free for general use. The publisher, the authors and the editors are safe to assume that the advice and information in this book are believed to be true and accurate at the date of publication. -

Inorganic Chemistry/Chemical Bonding/MO Diagram 1
Inorganic Chemistry/Chemical Bonding/MO Diagram 1 Inorganic Chemistry/Chemical Bonding/MO Diagram A molecular orbital diagram or MO diagram for short is a qualitative descriptive tool explaining chemical bonding in molecules in terms of molecular orbital theory in general and the Linear combination of atomic orbitals molecular orbital method (LCAO method) in particular [1] [2] [3] . This tool is very well suited for simple diatomic molecules such as dihydrogen, dioxygen and carbon monoxide but becomes more complex when discussing polynuclear molecules such as methane. It explains why some molecules exist and not others, how strong bonds are, and what electronic transitions take place. Dihydrogen MO diagram The smallest molecule, hydrogen gas exists as dihydrogen (H-H) with a single covalent bond between two hydrogen atoms. As each hydrogen atom has a single 1s atomic orbital for its electron, the bond forms by overlap of these two atomic orbitals. In figure 1 the two atomic orbitals are depicted on the left and on the right. The vertical axis always represents the orbital energies. The atomic orbital energy correlates with electronegativity as a more electronegative atom holds an electron more tightly thus lowering its energy. MO treatment is only valid when the atomic orbitals have comparable energy; when they differ greatly the mode of bonding becomes ionic. Each orbital is singly occupied with the up and down arrows representing an electron. The two AO's can overlap in two ways depending on their phase relationship. The phase of an orbital is a direct consequence of the oscillating, wave-like properties of electrons. -

Calculating Lattice Energies Using the Born-Haber Cycle an Extension Activity for AP Chemistry Students
Calculating Lattice Energies Using the Born-Haber Cycle An Extension Activity for AP Chemistry Students A particular set of equations known as a Born-Haber cycle demonstrates how chemists are able to use the first law of thermodynamics—that the energy of the universe is conserved in any chemical or physical change—to find an unknown energy value that is difficult or impossible to measure experimentally. Some energy quantities, such as the lattice energy of a mineral or the electron affinity of an atom, can be difficult to measure in the lab. Examining a Born-Haber cycle we see that there is more than one path to the formation of a substance in a particular state, and that if we use consistent definitions, an energy value that we seek can be calculated from energy values that we already know. The following exercise will help us see the way these energy values relate to one another, give us practice with their definitions and symbols, and deepen our insight to their meaning when we see them in other types of problems. Each physical or chemical change represented has: • an equation that represents a clearly defined physical or chemical change; • a definition of the particular type of energy change; • a symbol or abbreviation for the energy change equal to a value for the change in enthalpy (ΔH), the energy that is released or absorbed during the change, expressed in kJ/mol; and • a name by which that change in enthalpy is commonly known. To set up the equations in a Born-Haber cycle, cut out the cards for names, equations, defini- tions, and symbols with energy values. -

The Theoretical Study on Interaction of Hydrogen with Single-Walled Boron Nitride Nanotubes
THE JOURNAL OF CHEMICAL PHYSICS 123, 114703 ͑2005͒ The theoretical study on interaction of hydrogen with single-walled boron nitride nanotubes. I. The reactive force field ReaxFFHBN development ͒ Sang Soo Han, Jeung Ku Kang, and Hyuck Mo Leea Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea Adri C. T. van Duin and William A. Goddard III Materials and Process Simulation Center, California Institute of Technology, California 91125 ͑Received 2 November 2004; accepted 21 June 2005; published online 19 September 2005͒ We present a new reactive force field ReaxFFHBN derived to accurately model large molecular and condensed phase systems of H, B, and N atoms. ReaxFFHBN has been tested against quantum calculation data for B–H, B–B, and B–N bond dissociations and for H–B–H, B–N–B, and N–B–N bond angle strain energies of various molecular clusters. The accuracy of the developed ReaxFFHBN for B–N–H systems is also tested for ͑i͒ H–B and H–B bond energies as a function of out of plane ͑ ͒ ͑ ͒ ͑ ͒ in H–B NH2 3 and H–N BH2 3, respectively, ii the reaction energy for the B3N3H6 +H2 → ͑ ͒ B3N3H8, and iii crystal properties such as lattice parameters and equations of states for the hexagonal type ͑h-BN͒ with a graphite structure and for the cubic type ͑c-BN͒ with a zinc-blende structure. For all these systems, ReaxFFHBN gives reliable results consistent with those from quantum calculations as it describes well bond breaking and formation in chemical processes and physical properties. -

Session #9: Homework Solutions
Session #9: Homework Solutions Problem #1 Estimate the ionic radius of Cs+. The lattice energy of CsCl is 633 kJ/mol. For CsCl the Madelung constant, M, is 1.763, and the Born exponent, n, is 10.7. The ionic radius of Cl– is known to be 1.81 Å. Solution Mq q ⎛⎞1 E1 = 1 2 −= and r r+r lattice ⎜⎟ o Cs+ Cl− 4rπεoo⎝⎠ n Solve first for ro MqqN ⎛⎞1 1.763(1.6 x 10-19 ) 2 6.02 x 1023 ⎛ 1 ⎞ r1 = 12 Aν −− = 1 o ⎜⎟ -12 5 ⎜ ⎟ 4Eπεolattice⎝⎠ n 4π 8.85 x 10 6.33x10 ⎝10.7⎠ = 3.50 x 10-10 m = 3.50 Å = r+ r Cs+- Cl ∴ r = 3.50-1.81 = 1.69 Å Cs+ Problem #2 (a) CFCs have been implicated in ozone depletion. Show that when Freon 12 (CCl2F2) is exposed to ultraviolet radiation, the compound decomposes to produce chlorine. (b) Draw the Lewis structure of Freon 12 and indicate the polarities of each bond within this compound. (c) Determine the percent ionic character of the C--Cl and C--F bonds. DATA: Average Bond Energies (kJ/mol) Single Bonds Multiple Bonds H-H 435 C = C 610 F-F 155 C ≡ C 836 Cl-Cl 242 C-C 347 Solution (a) Chlorine will be liberated if the C-Cl bond breaks, therefore, compute its strength and show that ultraviolet photons have enough energy to break the bond. 2 ECCl−−− =+ E CCx9 E ClCl 6.3(χχC− Cl) = 347 x 242 + 96.3(2.55 - 3.16)2 = 326 x 103/6.02 x 1023 = 5.41 x 10-19 J/bond photon will break ∴this bond if Eph > Ebond ⇒ critical λ is hc 6.6 x 10-34 x 3 x 108 λ = = = 3.66 x 10-7 m -19 Ebond 5.41 x 10 which lies in the u.v. -

C – Laser 1. Introduction the Carbon Dioxide Laser (CO2 Laser) Was One
Generated by Foxit PDF Creator © Foxit Software http://www.foxitsoftware.com For evaluation only. C – laser 1. Introduction The carbon dioxide laser (CO2 laser) was one of the earliest gas lasers to be developed (invented by Kumar Patel of Bell Labs in 1964[1]), and is still one of the most useful. Carbon dioxide lasers are the highest-power continuous wave lasers that are currently available. They are also quite efficient: the ratio of output power to pump power can be as large as 20%. The CO2 laser produces a beam of infrared light with the principal wavelength bands centering around 9.4 and 10.6 micrometers. 2. Amplification The active laser medium is a gas discharge which is air-cooled (water-cooled in higher power applications). The filling gas within the discharge tube consists primarily of: o Carbon dioxide (CO2) (around 10–20%) o Nitrogen (N2) (around 10–20%) o Hydrogen (H2) and/or xenon (Xe) (a few percent; usually only used in a sealed tube.) o Helium (He) (The remainder of the gas mixture) The specific proportions vary according to the particular laser. The population inversion in the laser is achieved by the following sequence: 1) Electron impact excites vibrational motion of the nitrogen. Because nitrogen is a homonuclear molecule, it cannot lose this energy by photon emission, and its excited vibrational levels are therefore metastable and live for a long time. 2) Collisional energy transfer between the nitrogen and the carbon dioxide molecule causes vibrational excitation of the carbon dioxide, with sufficient efficiency to lead to the desired population inversion necessary for laser operation. -

Real Space in Situ Bond Energies: Toward a Consistent
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Repositorio Institucional de la Universidad de Oviedo Real space in situ bond energies: toward a consistent energetic definition of bond strength D. Menéndez-Crespo, A. Costales, E. Francisco, and A. Martín Pendása∗ June 20, 2018 Abstract A rigorous definition of intrinsic bond strength based on the partitioning of a molecule into real space fragments is presented. Using the domains provided by the quantum theory of atoms in molecules (QTAIM) together with the interacting quantum atoms (IQA) energetic decomposition, we show how an in situ bond strength, matching all the requirements of an intrinsic bond energy, can be defined between each pair of fragments. Total atomization or fragmentation energies are shown to be equal to the sum of these in situ bond energies (ISBEs) if the energies of the fragments are measured with respect to their in-the-molecule state. These energies usually lie above the ground state of the isolated fragments by quantities identified with the standard frag- ment relaxation or deformation energies, which are also provided by the protocol. Deformation energies bridge dissociation energies with ISBEs, and can be dissected using well-known tools of real space theories of chemical bonding. Similarly, ISBEs can be partitioned into ionic and covalent contributions, and this feature adds to the chemical appeal of the procedure. All the energetic quantities examined are observable and amenable, in principle, to experimental deter- mination. Several systems, exemplifying the role of each energetic term herein presented are used to show the power of the approach.