Introduction to Molecular Orbital Theory
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Direct Preparation of Some Organolithium Compounds from Lithium and RX Compounds Katashi Oita Iowa State College
Iowa State University Capstones, Theses and Retrospective Theses and Dissertations Dissertations 1955 Direct preparation of some organolithium compounds from lithium and RX compounds Katashi Oita Iowa State College Follow this and additional works at: https://lib.dr.iastate.edu/rtd Part of the Organic Chemistry Commons Recommended Citation Oita, Katashi, "Direct preparation of some organolithium compounds from lithium and RX compounds " (1955). Retrospective Theses and Dissertations. 14262. https://lib.dr.iastate.edu/rtd/14262 This Dissertation is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Retrospective Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. INFORMATION TO USERS This manuscript has been reproduced from the microfilm master. UMI films the text directly from the original or copy submitted. Thus, some thesis and dissertation copies are in typewriter face, while others may be from any type of computer printer. The quality of this reproduction is dependent upon the quality of the copy submitted. Broken or indistinct print, colored or poor quality illustrations and photographs, print bleedthrough, substandard margins, and improper alignment can adversely affect reproduction. In the unlikely event that the author did not send UMI a complete manuscript and there are missing pages, these will be noted. Also, if unauthorized copyright material had to be removed, a note will indicate the deletion. Oversize materials (e.g., maps, drawings, charts) are reproduced by sectioning the original, beginning at the upper left-hand comer and continuing from left to right in equal sections with small overiaps. -

Chemistry 2000 Slide Set 1: Introduction to the Molecular Orbital Theory
Chemistry 2000 Slide Set 1: Introduction to the molecular orbital theory Marc R. Roussel January 2, 2020 Marc R. Roussel Introduction to molecular orbitals January 2, 2020 1 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Hydrogenic atoms The hydrogenic atom (one nucleus, one electron) is exactly solvable. The solutions of this problem are called atomic orbitals. The square of the orbital wavefunction gives a probability density for the electron, i.e. the probability per unit volume of finding the electron near a particular point in space. Marc R. Roussel Introduction to molecular orbitals January 2, 2020 2 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Hydrogenic atoms (continued) Orbital shapes: 1s 2p 3dx2−y 2 3dz2 Marc R. Roussel Introduction to molecular orbitals January 2, 2020 3 / 24 Review: quantum mechanics of atoms Review: quantum mechanics of atoms Multielectron atoms Consider He, the simplest multielectron atom: Electron-electron repulsion makes it impossible to solve for the electronic wavefunctions exactly. A fourth quantum number, ms , which is associated with a new type of angular momentum called spin, enters into the theory. 1 1 For electrons, ms = 2 or − 2 . Pauli exclusion principle: No two electrons can have identical sets of quantum numbers. Consequence: Only two electrons can occupy an orbital. Marc R. Roussel Introduction to molecular orbitals January 2, 2020 4 / 24 The hydrogen molecular ion The quantum mechanics of molecules + H2 is the simplest possible molecule: two nuclei one electron Three-body problem: no exact solutions However, the nuclei are more than 1800 time heavier than the electron, so the electron moves much faster than the nuclei. -

Theoretical Methods That Help Understanding the Structure and Reactivity of Gas Phase Ions
International Journal of Mass Spectrometry 240 (2005) 37–99 Review Theoretical methods that help understanding the structure and reactivity of gas phase ions J.M. Merceroa, J.M. Matxaina, X. Lopeza, D.M. Yorkb, A. Largoc, L.A. Erikssond,e, J.M. Ugaldea,∗ a Kimika Fakultatea, Euskal Herriko Unibertsitatea, P.K. 1072, 20080 Donostia, Euskadi, Spain b Department of Chemistry, University of Minnesota, 207 Pleasant St. SE, Minneapolis, MN 55455-0431, USA c Departamento de Qu´ımica-F´ısica, Universidad de Valladolid, Prado de la Magdalena, 47005 Valladolid, Spain d Department of Cell and Molecular Biology, Box 596, Uppsala University, 751 24 Uppsala, Sweden e Department of Natural Sciences, Orebro¨ University, 701 82 Orebro,¨ Sweden Received 27 May 2004; accepted 14 September 2004 Available online 25 November 2004 Abstract The methods of the quantum electronic structure theory are reviewed and their implementation for the gas phase chemistry emphasized. Ab initio molecular orbital theory, density functional theory, quantum Monte Carlo theory and the methods to calculate the rate of complex chemical reactions in the gas phase are considered. Relativistic effects, other than the spin–orbit coupling effects, have not been considered. Rather than write down the main equations without further comments on how they were obtained, we provide the reader with essentials of the background on which the theory has been developed and the equations derived. We committed ourselves to place equations in their own proper perspective, so that the reader can appreciate more profoundly the subtleties of the theory underlying the equations themselves. Finally, a number of examples that illustrate the application of the theory are presented and discussed. -

Mineral Processing
Mineral Processing Foundations of theory and practice of minerallurgy 1st English edition JAN DRZYMALA, C. Eng., Ph.D., D.Sc. Member of the Polish Mineral Processing Society Wroclaw University of Technology 2007 Translation: J. Drzymala, A. Swatek Reviewer: A. Luszczkiewicz Published as supplied by the author ©Copyright by Jan Drzymala, Wroclaw 2007 Computer typesetting: Danuta Szyszka Cover design: Danuta Szyszka Cover photo: Sebastian Bożek Oficyna Wydawnicza Politechniki Wrocławskiej Wybrzeze Wyspianskiego 27 50-370 Wroclaw Any part of this publication can be used in any form by any means provided that the usage is acknowledged by the citation: Drzymala, J., Mineral Processing, Foundations of theory and practice of minerallurgy, Oficyna Wydawnicza PWr., 2007, www.ig.pwr.wroc.pl/minproc ISBN 978-83-7493-362-9 Contents Introduction ....................................................................................................................9 Part I Introduction to mineral processing .....................................................................13 1. From the Big Bang to mineral processing................................................................14 1.1. The formation of matter ...................................................................................14 1.2. Elementary particles.........................................................................................16 1.3. Molecules .........................................................................................................18 1.4. Solids................................................................................................................19 -

Valence Bond Theory
UMass Boston, Chem 103, Spring 2006 CHEM 103 Molecular Geometry and Valence Bond Theory Lecture Notes May 2, 2006 Prof. Sevian Announcements z The final exam is scheduled for Monday, May 15, 8:00- 11:00am It will NOT be in our regularly scheduled lecture hall (S- 1-006). The final exam location has been changed to Snowden Auditorium (W-1-088). © 2006 H. Sevian 1 UMass Boston, Chem 103, Spring 2006 More announcements Information you need for registering for the second semester of general chemistry z If you will take it in the summer: z Look for chem 104 in the summer schedule (includes lecture and lab) z If you will take it in the fall: z Look for chem 116 (lecture) and chem 118 (lab). These courses are co-requisites. z If you plan to re-take chem 103, in the summer it will be listed as chem 103 (lecture + lab). In the fall it will be listed as chem 115 (lecture) + chem 117 (lab), which are co-requisites. z Note: you are only eligible for a lab exemption if you previously passed the course. Agenda z Results of Exam 3 z Molecular geometries observed z How Lewis structure theory predicts them z Valence shell electron pair repulsion (VSEPR) theory z Valence bond theory z Bonds are formed by overlap of atomic orbitals z Before atoms bond, their atomic orbitals can hybridize to prepare for bonding z Molecular geometry arises from hybridization of atomic orbitals z σ and π bonding orbitals © 2006 H. Sevian 2 UMass Boston, Chem 103, Spring 2006 Molecular Geometries Observed Tetrahedral See-saw Square planar Square pyramid Lewis Structure Theory -

Lewis Structure Handout
Lewis Structure Handout for Chemistry Students An electron dot diagram, also known as a Lewis Structure, is a representation of valence electrons in a single atom, and can be further utilized to depict the bonds that form based on the available electrons for covalent bonding between multiple atoms. -Each atom has a characteristic dot diagram based on its position in the periodic table and this reflects its potential for fulfillment of the octet rule. As a general rule, the noble gases have a filled octet (column VIII with eight valence electrons) and each preceding column has successively one fewer. • For example, Carbon is in column IV and has four valence electrons. It can therefore be represented as: • Each of these "lone" electrons can form a bond by sharing the orbital with another unbonded electron. Hydrogen, being in column I, has a single valence electron, and will therefore satisfy carbon's octet thusly: • When a compound has bonded electrons (as with CH4) it is most accurate to diagram it with lines representing each of the single bonds. • Using carbon again for a slightly more complicated example, it can also form double bonds. This is the case when carbon bonds to two oxygen atoms, resulting in CO2. The oxygen atoms (with six valence electrons in column VI) are each represented as: • Because carbon is the least electronegative of the atoms involved, it is placed centrally, and its valence electrons will migrate toward the more electronegative oxygens to form two double bonds and a linear structure. This can be written as: where oxygen's remaining lone pairs are still shown. -

Molecular Orbital Theory to Predict Bond Order • to Apply Molecular Orbital Theory to the Diatomic Homonuclear Molecule from the Elements in the Second Period
Skills to Develop • To use molecular orbital theory to predict bond order • To apply Molecular Orbital Theory to the diatomic homonuclear molecule from the elements in the second period. None of the approaches we have described so far can adequately explain why some compounds are colored and others are not, why some substances with unpaired electrons are stable, and why others are effective semiconductors. These approaches also cannot describe the nature of resonance. Such limitations led to the development of a new approach to bonding in which electrons are not viewed as being localized between the nuclei of bonded atoms but are instead delocalized throughout the entire molecule. Just as with the valence bond theory, the approach we are about to discuss is based on a quantum mechanical model. Previously, we described the electrons in isolated atoms as having certain spatial distributions, called orbitals, each with a particular orbital energy. Just as the positions and energies of electrons in atoms can be described in terms of atomic orbitals (AOs), the positions and energies of electrons in molecules can be described in terms of molecular orbitals (MOs) A particular spatial distribution of electrons in a molecule that is associated with a particular orbital energy.—a spatial distribution of electrons in a molecule that is associated with a particular orbital energy. As the name suggests, molecular orbitals are not localized on a single atom but extend over the entire molecule. Consequently, the molecular orbital approach, called molecular orbital theory is a delocalized approach to bonding. Although the molecular orbital theory is computationally demanding, the principles on which it is based are similar to those we used to determine electron configurations for atoms. -

8.3 Bonding Theories >
8.3 Bonding Theories > Chapter 8 Covalent Bonding 8.1 Molecular Compounds 8.2 The Nature of Covalent Bonding 8.3 Bonding Theories 8.4 Polar Bonds and Molecules 1 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Molecular Orbitals How are atomic and molecular orbitals related? 2 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals • The model you have been using for covalent bonding assumes the orbitals are those of the individual atoms. • There is a quantum mechanical model of bonding, however, that describes the electrons in molecules using orbitals that exist only for groupings of atoms. 3 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals • When two atoms combine, this model assumes that their atomic orbitals overlap to produce molecular orbitals, or orbitals that apply to the entire molecule. 4 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Just as an atomic orbital belongs to a particular atom, a molecular orbital belongs to a molecule as a whole. • A molecular orbital that can be occupied by two electrons of a covalent bond is called a bonding orbital. 5 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Sigma Bonds When two atomic orbitals combine to form a molecular orbital that is symmetrical around the axis connecting two atomic nuclei, a sigma bond is formed. • Its symbol is the Greek letter sigma (σ). -
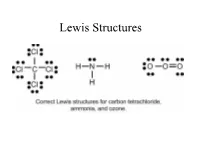
Lewis Structures
Lewis Structures Valence electrons for Elements Recall that the valence electrons for the elements can be determined based on the elements position on the periodic table. Lewis Dot Symbol Valence electrons and number of bonds Number of bonds elements prefers depending on the number of valence electrons. In general - F a m i l y → # C o v a l e n t B o n d s* H a l o g e n s X F , B r , C l , I → 1 bond often C a l c o g e n s O 2 bond often O , S → N i t r o g e n N 3 bond often N , P → C a r b o n C → 4 bond always C , S i The above chart is a guide on the number of bonds formed by these atoms. Lewis Structure, Octet Rule Guidelines When compounds are formed they tend to follow the Octet Rule. Octet Rule: Atoms will share electrons (e-) until it is surrounded by eight valence electrons. 4 unpaired 3unpaired 2unpaired 1unpaired up = unpaired e- 4 bonds 3 bonds 2 bonds 1 bond O=C=O N≡ N O = O F - F Atomic Connectivity The atomic arrangement for a molecule is usually given. CH2ClF HNO3 CH3COOH H2SO4 Cl N H O O O O H C F O H C C H O S O H H H H O H O In general when there is a single central atom in the 3- molecule, CH2ClF, SeCl2, O3 (CO2, NH3, PO4 ), the central atom is the first atom in the chemical formula. -

Draw Three Resonance Structures for the Chlorate Ion, Clo3
University Chemistry Quiz 3 2014/11/6 + 1. (10%) Explain why the bond order of N2 is greater than that of N2 , but the bond + order of O2 is less than that of O2 . Sol. + In forming the N2 from N2, an electron is removed from the sigma bonding molecular orbital. Consequently, the bond order decreases to 2.5 from 3.0. In + forming the O2 ion from O2, an electron is removed from the pi antibonding molecular orbital. Consequently, the bond order increases to 2.5 from 2.0. - 2. (10%) Draw three resonance structures for the chlorate ion, ClO3 . Show formal charges. Sol. Strategy: We follow the procedure for drawing Lewis structures outlined in Section 3.4 of the text. After we complete the Lewis structure, we draw the resonance structures. Solution: Following the procedure in Section 3.4 of the text, we come up with − the following Lewis structure for ClO3 . O − + − O Cl O We can draw two more equivalent Lewis structures with the double bond between Cl and a different oxygen atom. The resonance structures with formal charges are as follows: − − O O O + − − + − − + O Cl O O Cl O O Cl O 3. (5%) Use the molecular orbital energy-level diagram for O2 to show that the following Lewis structure corresponds to an excited state: Sol. The Lewis structure shows 4 pairs of electrons on the two oxygen atoms. From Table 3.4 of the text, we see that these 8 valence electrons are placed in the σ p p p p 2p , 2p , 2p , 2p , and 2p orbitals. -

Molecular Orbital Theory
Molecular Orbital Theory The Lewis Structure approach provides an extremely simple method for determining the electronic structure of many molecules. It is a bit simplistic, however, and does have trouble predicting structures for a few molecules. Nevertheless, it gives a reasonable structure for many molecules and its simplicity to use makes it a very useful tool for chemists. A more general, but slightly more complicated approach is the Molecular Orbital Theory. This theory builds on the electron wave functions of Quantum Mechanics to describe chemical bonding. To understand MO Theory let's first review constructive and destructive interference of standing waves starting with the full constructive and destructive interference that occurs when standing waves overlap completely. When standing waves only partially overlap we get partial constructive and destructive interference. To see how we use these concepts in Molecular Orbital Theory, let's start with H2, the simplest of all molecules. The 1s orbitals of the H-atom are standing waves of the electron wavefunction. In Molecular Orbital Theory we view the bonding of the two H-atoms as partial constructive interference between standing wavefunctions of the 1s orbitals. The energy of the H2 molecule with the two electrons in the bonding orbital is lower by 435 kJ/mole than the combined energy of the two separate H-atoms. On the other hand, the energy of the H2 molecule with two electrons in the antibonding orbital is higher than two separate H-atoms. To show the relative energies we use diagrams like this: In the H2 molecule, the bonding and anti-bonding orbitals are called sigma orbitals (σ). -

Chemical Bonding II: Molecular Shapes, Valence Bond Theory, and Molecular Orbital Theory Review Questions
Chemical Bonding II: Molecular Shapes, Valence Bond Theory, and Molecular Orbital Theory Review Questions 10.1 J The properties of molecules are directly related to their shape. The sensation of taste, immune response, the sense of smell, and many types of drug action all depend on shape-specific interactions between molecules and proteins. According to VSEPR theory, the repulsion between electron groups on interior atoms of a molecule determines the geometry of the molecule. The five basic electron geometries are (1) Linear, which has two electron groups. (2) Trigonal planar, which has three electron groups. (3) Tetrahedral, which has four electron groups. (4) Trigonal bipyramid, which has five electron groups. (5) Octahedral, which has six electron groups. An electron group is defined as a lone pair of electrons, a single bond, a multiple bond, or even a single electron. H—C—H 109.5= ijj^^jl (a) Linear geometry \ \ (b) Trigonal planar geometry I Tetrahedral geometry I Equatorial chlorine Axial chlorine "P—Cl: \ Trigonal bipyramidal geometry 1 I Octahedral geometry I 369 370 Chapter 10 Chemical Bonding II The electron geometry is the geometrical arrangement of the electron groups around the central atom. The molecular geometry is the geometrical arrangement of the atoms around the central atom. The electron geometry and the molecular geometry are the same when every electron group bonds two atoms together. The presence of unbonded lone-pair electrons gives a different molecular geometry and electron geometry. (a) Four electron groups give tetrahedral electron geometry, while three bonding groups and one lone pair give a trigonal pyramidal molecular geometry.