Simple Molecular Orbitals - Sigma and Pi Bonds in Molecules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

No Rabbit Ears on Water. the Structure of the Water Molecule
No Rabbit Ears on Water The Structure of the Water Molecule: What Should We Tell the Students? Michael Laing University of Natal. Durban, South Africa In the vast majority of high school (I)and freshman uni- He also considers the possibility of s character in the bond- versity (2) chemistry textbooks, water is presented as a bent ing orbitals of OF2 and OH2 to improve the "strength" of the molecule whose bonding can be described bv the Gilles~ie- orbital (p 111) from 1.732 for purep to 2.00 for the ideal sp3. Nyholm approach with the oxygen atomsp3 Gyhridized. The Inclusion of s character in the hybrid will be "resisted in the lone uairs are said to he dominant causine the H-O-H anele case of atoms with an nnshared pair.. in the s orbital, to beieduced from the ideal 1091120to 104' and are regul&ly which is more stable than the p orbitals" (p 120). Estimates shown as equivalent in shape and size even being described of the H-E-H angle range from 91.2 for AsH3 to 93.5O for as "rabbit ears" (3). Chemists have even been advised to Hz0. He once again ascribes the anomalies for Hz0 to "re- wear Mickey Mouse ears when teaching the structure and uulsion of the charzes of the hvdroaeu atoms" (D 122). bonding of the H20molecule, presumably to emphasize the - In his famous hook (6)~itac~oro&kii describks the bond- shape of the molecule (and possibly the two equivalent lone ing in water and HzS (p 10). -

Transcription 12.02.15
Lecture 12A • 02/15/12 We’re going to continue our discussion of conjugation. If we go back and talk about electrons and how they act like waves, [there’s] something known as the particle in a box. You’ve got some kind of box where imagine that the walls are infinitely tall. It’s a one-dimensional system, where the electron, all it can do, is go left and right between the two walls of the box. The electron’s a wave; it can’t exist outside the box, so whatever function, whatever wave we use to describe the electron, has to have a value of zero at one end of the box and zero at the other end of the box. Graphically, what do the solutions look like? We have something like this, where we’re talking about energy potential; it’s infinite at either end of the box, and zero in between. We have an electron that’s bouncing around inside of it. An electron’s a wave, and there’s function that describes that wave. The only way it can fit in this box and physically make sense is if the wave starts and stops and the ends of the box. It turns out that – long, long, long story short – one of the solutions for the Schrödinger equation is just a sine function – actually, it’s part of an exponential version of a sine function. It’s something, at least, we can draw a pretty picture of. That’s why you’ll start with this problem in a discussion of quantum mechanics, because it is solvable, it is only on in dimension, and it’s more humanly possible to discuss. -

Aromaticity Sem- Ii
AROMATICITY SEM- II In 1931, German chemist and physicist Sir Erich Hückel proposed a theory to help determine if a planar ring molecule would have aromatic properties .This is a very popular and useful rule to identify aromaticity in monocyclic conjugated compound. According to which a planar monocyclic conjugated system having ( 4n +2) delocalised (where, n = 0, 1, 2, .....) electrons are known as aromatic compound . For example: Benzene, Naphthalene, Furan, Pyrrole etc. Criteria for Aromaticity 1) The molecule is cyclic (a ring of atoms) 2) The molecule is planar (all atoms in the molecule lie in the same plane) 3) The molecule is fully conjugated (p orbitals at every atom in the ring) 4) The molecule has 4n+2 π electrons (n=0 or any positive integer Why 4n+2π Electrons? According to Hückel's Molecular Orbital Theory, a compound is particularly stable if all of its bonding molecular orbitals are filled with paired electrons. - This is true of aromatic compounds, meaning they are quite stable. - With aromatic compounds, 2 electrons fill the lowest energy molecular orbital, and 4 electrons fill each subsequent energy level (the number of subsequent energy levels is denoted by n), leaving all bonding orbitals filled and no anti-bonding orbitals occupied. This gives a total of 4n+2π electrons. - As for example: Benzene has 6π electrons. Its first 2π electrons fill the lowest energy orbital, and it has 4π electrons remaining. These 4 fill in the orbitals of the succeeding energy level. The criteria for Antiaromaticity are as follows: 1) The molecule must be cyclic and completely conjugated 2) The molecule must be planar. -
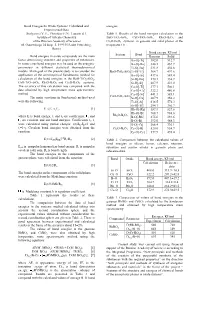
Bond Energies in Oxide Systems: Calculated and Experimental Data
Bond Energies In Oxide Systems: Calculated and energies. Experimental Data Stolyarova V. L., Plotnikov E.N., Lopatin S.I. Table 1. Results of the bond energies calculation in the Institute of Silicate Chemistry BaO-TiO 2-SiO 2, CaO-TiO 2-SiO 2, Rb 2O-B2O3 and of the Russian Academy of Sciences Cs 2O-B2O3 systems. in gaseous and solid phases at the ul. Odoevskogo 24 korp. 2, 199155 Sankt Petersburg, temperature 0 ¥ . Russia Bond energy, KJ/mol System Bond Bond energies in oxide compounds are the main Gaseous Solid factor determining structure and properties of substances. Ba-O[-Ti] 382.0 512.7 In some cases bond energies may be used as the energetic Ba-O[-Ba] 348.5 467.7 parameters in different statistical thermodynamical Ti-O[-Ba] 331.8 450.6 models. Main goal of the present study is to consider the BaO-TiO 2-SiO 2 Ti-O[-Ti] 364.7 497.6 application of the semiempirical Sanderson's method for Ba-O[-Si] 437.6 585.4 calculation of the bond energies in the BaO-TiO 2-SiO 2, Si-O[-Ba] 270.3 334.7 CaO-TiO 2-SiO 2, Rb 2O-B2O3 and Cs 2O-B2O3 systems. Si-O[-Si] 407.9 421.0 The accuracy of this calculation was compared with the Ca-O[-Ti] 377.1 500.1 data obtained by high temperature mass spectrometric Ca-O[-Ca] 322.2 486.8 method. Ca-O[-Si] 441.1 585.1 CaO-TiO -SiO The main equations in Sanderson's method used 2 2 Si-O[-Ca] 287.9 358.2 were the following. -

Molecular Orbital Theory to Predict Bond Order • to Apply Molecular Orbital Theory to the Diatomic Homonuclear Molecule from the Elements in the Second Period
Skills to Develop • To use molecular orbital theory to predict bond order • To apply Molecular Orbital Theory to the diatomic homonuclear molecule from the elements in the second period. None of the approaches we have described so far can adequately explain why some compounds are colored and others are not, why some substances with unpaired electrons are stable, and why others are effective semiconductors. These approaches also cannot describe the nature of resonance. Such limitations led to the development of a new approach to bonding in which electrons are not viewed as being localized between the nuclei of bonded atoms but are instead delocalized throughout the entire molecule. Just as with the valence bond theory, the approach we are about to discuss is based on a quantum mechanical model. Previously, we described the electrons in isolated atoms as having certain spatial distributions, called orbitals, each with a particular orbital energy. Just as the positions and energies of electrons in atoms can be described in terms of atomic orbitals (AOs), the positions and energies of electrons in molecules can be described in terms of molecular orbitals (MOs) A particular spatial distribution of electrons in a molecule that is associated with a particular orbital energy.—a spatial distribution of electrons in a molecule that is associated with a particular orbital energy. As the name suggests, molecular orbitals are not localized on a single atom but extend over the entire molecule. Consequently, the molecular orbital approach, called molecular orbital theory is a delocalized approach to bonding. Although the molecular orbital theory is computationally demanding, the principles on which it is based are similar to those we used to determine electron configurations for atoms. -

Introduction to Molecular Orbital Theory
Chapter 2: Molecular Structure and Bonding Bonding Theories 1. VSEPR Theory 2. Valence Bond theory (with hybridization) 3. Molecular Orbital Theory ( with molecualr orbitals) To date, we have looked at three different theories of molecular boning. They are the VSEPR Theory (with Lewis Dot Structures), the Valence Bond theory (with hybridization) and Molecular Orbital Theory. A good theory should predict physical and chemical properties of the molecule such as shape, bond energy, bond length, and bond angles.Because arguments based on atomic orbitals focus on the bonds formed between valence electrons on an atom, they are often said to involve a valence-bond theory. The valence-bond model can't adequately explain the fact that some molecules contains two equivalent bonds with a bond order between that of a single bond and a double bond. The best it can do is suggest that these molecules are mixtures, or hybrids, of the two Lewis structures that can be written for these molecules. This problem, and many others, can be overcome by using a more sophisticated model of bonding based on molecular orbitals. Molecular orbital theory is more powerful than valence-bond theory because the orbitals reflect the geometry of the molecule to which they are applied. But this power carries a significant cost in terms of the ease with which the model can be visualized. One model does not describe all the properties of molecular bonds. Each model desribes a set of properties better than the others. The final test for any theory is experimental data. Introduction to Molecular Orbital Theory The Molecular Orbital Theory does a good job of predicting elctronic spectra and paramagnetism, when VSEPR and the V-B Theories don't. -

8.3 Bonding Theories >
8.3 Bonding Theories > Chapter 8 Covalent Bonding 8.1 Molecular Compounds 8.2 The Nature of Covalent Bonding 8.3 Bonding Theories 8.4 Polar Bonds and Molecules 1 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Molecular Orbitals How are atomic and molecular orbitals related? 2 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals • The model you have been using for covalent bonding assumes the orbitals are those of the individual atoms. • There is a quantum mechanical model of bonding, however, that describes the electrons in molecules using orbitals that exist only for groupings of atoms. 3 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals • When two atoms combine, this model assumes that their atomic orbitals overlap to produce molecular orbitals, or orbitals that apply to the entire molecule. 4 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Just as an atomic orbital belongs to a particular atom, a molecular orbital belongs to a molecule as a whole. • A molecular orbital that can be occupied by two electrons of a covalent bond is called a bonding orbital. 5 Copyright © Pearson Education, Inc., or its affiliates. All Rights Reserved. 8.3 Bonding Theories > Molecular Orbitals Sigma Bonds When two atomic orbitals combine to form a molecular orbital that is symmetrical around the axis connecting two atomic nuclei, a sigma bond is formed. • Its symbol is the Greek letter sigma (σ). -

Molecular Orbital Theory
Molecular Orbital Theory The Lewis Structure approach provides an extremely simple method for determining the electronic structure of many molecules. It is a bit simplistic, however, and does have trouble predicting structures for a few molecules. Nevertheless, it gives a reasonable structure for many molecules and its simplicity to use makes it a very useful tool for chemists. A more general, but slightly more complicated approach is the Molecular Orbital Theory. This theory builds on the electron wave functions of Quantum Mechanics to describe chemical bonding. To understand MO Theory let's first review constructive and destructive interference of standing waves starting with the full constructive and destructive interference that occurs when standing waves overlap completely. When standing waves only partially overlap we get partial constructive and destructive interference. To see how we use these concepts in Molecular Orbital Theory, let's start with H2, the simplest of all molecules. The 1s orbitals of the H-atom are standing waves of the electron wavefunction. In Molecular Orbital Theory we view the bonding of the two H-atoms as partial constructive interference between standing wavefunctions of the 1s orbitals. The energy of the H2 molecule with the two electrons in the bonding orbital is lower by 435 kJ/mole than the combined energy of the two separate H-atoms. On the other hand, the energy of the H2 molecule with two electrons in the antibonding orbital is higher than two separate H-atoms. To show the relative energies we use diagrams like this: In the H2 molecule, the bonding and anti-bonding orbitals are called sigma orbitals (σ). -

Inorganic Chemistry/Chemical Bonding/MO Diagram 1
Inorganic Chemistry/Chemical Bonding/MO Diagram 1 Inorganic Chemistry/Chemical Bonding/MO Diagram A molecular orbital diagram or MO diagram for short is a qualitative descriptive tool explaining chemical bonding in molecules in terms of molecular orbital theory in general and the Linear combination of atomic orbitals molecular orbital method (LCAO method) in particular [1] [2] [3] . This tool is very well suited for simple diatomic molecules such as dihydrogen, dioxygen and carbon monoxide but becomes more complex when discussing polynuclear molecules such as methane. It explains why some molecules exist and not others, how strong bonds are, and what electronic transitions take place. Dihydrogen MO diagram The smallest molecule, hydrogen gas exists as dihydrogen (H-H) with a single covalent bond between two hydrogen atoms. As each hydrogen atom has a single 1s atomic orbital for its electron, the bond forms by overlap of these two atomic orbitals. In figure 1 the two atomic orbitals are depicted on the left and on the right. The vertical axis always represents the orbital energies. The atomic orbital energy correlates with electronegativity as a more electronegative atom holds an electron more tightly thus lowering its energy. MO treatment is only valid when the atomic orbitals have comparable energy; when they differ greatly the mode of bonding becomes ionic. Each orbital is singly occupied with the up and down arrows representing an electron. The two AO's can overlap in two ways depending on their phase relationship. The phase of an orbital is a direct consequence of the oscillating, wave-like properties of electrons. -

Covalent Bonding and Molecular Orbitals
Covalent Bonding and Molecular Orbitals Chemistry 35 Fall 2000 From Atoms to Molecules: The Covalent Bond n So, what happens to e- in atomic orbitals when two atoms approach and form a covalent bond? Mathematically: -let’s look at the formation of a hydrogen molecule: -we start with: 1 e-/each in 1s atomic orbitals -we’ll end up with: 2 e- in molecular obital(s) HOW? Make linear combinations of the 1s orbital wavefunctions: ymol = y1s(A) ± y1s(B) Then, solve via the SWE! 2 1 Hydrogen Wavefunctions wavefunctions probability densities 3 Hydrogen Molecular Orbitals anti-bonding bonding 4 2 Hydrogen MO Formation: Internuclear Separation n SWE solved with nuclei at a specific separation distance . How does the energy of the new MO vary with internuclear separation? movie 5 MO Theory: Homonuclear Diatomic Molecules n Let’s look at the s-bonding properties of some homonuclear diatomic molecules: Bond order = ½(bonding e- - anti-bonding e-) For H2: B.O. = 1 - 0 = 1 (single bond) For He2: B.O. = 1 - 1 = 0 (no bond) 6 3 Configurations and Bond Orders: 1st Period Diatomics Species Config. B.O. Energy Length + 1 H2 (s1s) ½ 255 kJ/mol 1.06 Å 2 H2 (s1s) 1 431 kJ/mol 0.74 Å + 2 * 1 He2 (s1s) (s 1s) ½ 251 kJ/mol 1.08 Å 2 * 2 He2 (s1s) (s 1s) 0 ~0 LARGE 7 Combining p-orbitals: s and p MO’s antibonding end-on overlap bonding antibonding side-on overlap bonding antibonding bonding8 4 2nd Period MO Energies s2p has lowest energy due to better overlap (end-on) of 2pz orbitals p2p orbitals are degenerate and at higher energy than the s2p 9 2nd Period MO Energies: Shift! For Z <8: 2s and 2p orbitals can interact enough to change energies of the resulting s2s and s2p MOs. -

Calculating Lattice Energies Using the Born-Haber Cycle an Extension Activity for AP Chemistry Students
Calculating Lattice Energies Using the Born-Haber Cycle An Extension Activity for AP Chemistry Students A particular set of equations known as a Born-Haber cycle demonstrates how chemists are able to use the first law of thermodynamics—that the energy of the universe is conserved in any chemical or physical change—to find an unknown energy value that is difficult or impossible to measure experimentally. Some energy quantities, such as the lattice energy of a mineral or the electron affinity of an atom, can be difficult to measure in the lab. Examining a Born-Haber cycle we see that there is more than one path to the formation of a substance in a particular state, and that if we use consistent definitions, an energy value that we seek can be calculated from energy values that we already know. The following exercise will help us see the way these energy values relate to one another, give us practice with their definitions and symbols, and deepen our insight to their meaning when we see them in other types of problems. Each physical or chemical change represented has: • an equation that represents a clearly defined physical or chemical change; • a definition of the particular type of energy change; • a symbol or abbreviation for the energy change equal to a value for the change in enthalpy (ΔH), the energy that is released or absorbed during the change, expressed in kJ/mol; and • a name by which that change in enthalpy is commonly known. To set up the equations in a Born-Haber cycle, cut out the cards for names, equations, defini- tions, and symbols with energy values. -

The Theoretical Study on Interaction of Hydrogen with Single-Walled Boron Nitride Nanotubes
THE JOURNAL OF CHEMICAL PHYSICS 123, 114703 ͑2005͒ The theoretical study on interaction of hydrogen with single-walled boron nitride nanotubes. I. The reactive force field ReaxFFHBN development ͒ Sang Soo Han, Jeung Ku Kang, and Hyuck Mo Leea Department of Materials Science and Engineering, Korea Advanced Institute of Science and Technology (KAIST), Daejeon 305-701, Republic of Korea Adri C. T. van Duin and William A. Goddard III Materials and Process Simulation Center, California Institute of Technology, California 91125 ͑Received 2 November 2004; accepted 21 June 2005; published online 19 September 2005͒ We present a new reactive force field ReaxFFHBN derived to accurately model large molecular and condensed phase systems of H, B, and N atoms. ReaxFFHBN has been tested against quantum calculation data for B–H, B–B, and B–N bond dissociations and for H–B–H, B–N–B, and N–B–N bond angle strain energies of various molecular clusters. The accuracy of the developed ReaxFFHBN for B–N–H systems is also tested for ͑i͒ H–B and H–B bond energies as a function of out of plane ͑ ͒ ͑ ͒ ͑ ͒ in H–B NH2 3 and H–N BH2 3, respectively, ii the reaction energy for the B3N3H6 +H2 → ͑ ͒ B3N3H8, and iii crystal properties such as lattice parameters and equations of states for the hexagonal type ͑h-BN͒ with a graphite structure and for the cubic type ͑c-BN͒ with a zinc-blende structure. For all these systems, ReaxFFHBN gives reliable results consistent with those from quantum calculations as it describes well bond breaking and formation in chemical processes and physical properties.