Abstract-Book
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Analysis of Short Term Precipitation in Norway 1967-2010
no. 15/2011 Climate ANALYSIS OF SHORT TERM PRECIPITATION IN NORWAY 1967-2010 Jostein Mamen, Rasmus Benestad and Jan Erik Haugen Photo: Einar Egeland . Contents 1 Introduction 4 1.1 Observation of short term precipitation in Norway . 4 1.2 Types of stations . 4 1.2.1 Tipping bucket rain gauge stations . 4 1.2.2 Weight pluviometer stations . 4 2 Rainfall rate statistics from tipping bucket rain gauge data 5 2.1 Maximum recorded 1 minute values . 5 2.2 Seasonal variations . 8 3 Rainfall rate statistics from weight pluviometer data 10 3.1 Maximum recorded 1 hour values . 10 3.2 Seasonal variations . 12 3.3 Conversion method from 1 hour to 1 minute precipitation . 12 4 Rainfall rate maps 13 4.1 Rainfall intensity exceeded 0.001 % of the time . 13 4.2 Rainfall intensity exceeded 0.01 % of the time . 16 4.2.1 Monthly values . 19 4.3 Rainfall intensity exceeded 0.1 % of the time . 19 4.4 Rainfall intensity exceeded 0.5 % of the time . 22 5 Rainfall rate trends from tipping bucket data and return periods 24 5.1 Rainfall rate trends . 24 5.1.1 Trends of maximum annual 1-minute intensity . 24 5.1.2 Trends of intensity exceeded 0.01 % of the time . 24 5.2 Return periods . 24 6 Methods used to derive rainfall intensity data from long term historical data set 26 7 Appendix - list of stations 33 3 1 Introduction In telecommunication heavy precipitation can lead to outage. [1], [4]. Met.no is therefore asked to analyze short term precipitation in Norway, down to one minute's time resolution. -

A Bid for Better Transit Improving Service with Contracted Operations Transitcenter Is a Foundation That Works to Improve Urban Mobility
A Bid for Better Transit Improving service with contracted operations TransitCenter is a foundation that works to improve urban mobility. We believe that fresh thinking can change the transportation landscape and improve the overall livability of cities. We commission and conduct research, convene events, and produce publications that inform and improve public transit and urban transportation. For more information, please visit www.transitcenter.org. The Eno Center for Transportation is an independent, nonpartisan think tank that promotes policy innovation and leads professional development in the transportation industry. As part of its mission, Eno seeks continuous improvement in transportation and its public and private leadership in order to improve the system’s mobility, safety, and sustainability. For more information please visit: www.enotrans.org. TransitCenter Board of Trustees Rosemary Scanlon, Chair Eric S. Lee Darryl Young Emily Youssouf Jennifer Dill Clare Newman Christof Spieler A Bid for Better Transit Improving service with contracted operations TransitCenter + Eno Center for Transportation September 2017 Acknowledgments A Bid for Better Transit was written by Stephanie Lotshaw, Paul Lewis, David Bragdon, and Zak Accuardi. The authors thank Emily Han, Joshua Schank (now at LA Metro), and Rob Puentes of the Eno Center for their contributions to this paper’s research and writing. This report would not be possible without the dozens of case study interviewees who contributed their time and knowledge to the study and reviewed the report’s case studies (see report appendices). The authors are also indebted to Don Cohen, Didier van de Velde, Darnell Grisby, Neil Smith, Kent Woodman, Dottie Watkins, Ed Wytkind, and Jeff Pavlak for their detailed and insightful comments during peer review. -

Vehicle Routing Problem Applied for Demand Controlled Waste Collection
Anett Cammermeyer Katrine Lunde Master Thesis - Vehicle routing problem applied for demand controlled waste collection - GRA 19003 Master of Science in Business and Economics: Logistics – Supply Chains and Networks Supervisor: Mehdi Sharifyazdi Date of Submission BI Norwegian Business School, Oslo Deadline 29.08.2014 01.09.2014 “This thesis is a part of the MSc Program at BI Norwegian Business School. The School takes no responsibility for the methods used, results found and conclusions drawn” GRA 19003 Master Thesis 01.09.2014 Acknowledgement This thesis is a submission to BI Norwegian Business School and completes our MSc degree in Logistics – Supply Chains and Networks, and thereby rounds out our five-year long education. The process of writing this thesis has been challenging, however interesting. We have learned a lot and know to this day that this is an experience we would not be without. We would like to thank Renovasjonsetaten and Sørum, which provided us with some necessary data needed for this thesis and giving us this opportunity. We would also like to give a special thanks to the chauffeur who let us participate on a route, and provided us with a lot of interesting information needed to understand the complexity of the work. The project has been very challenging and we would not have made it without the help of our supervisor Mehdi Sharifyazdi. His competence, guidance, time and insightful feedback have been a huge part of this thesis. At the end we will like to thank our partners and family for good support and positive enthusiasm during the work with this Master Thesis. -

Og Bygningsetaten Planforslag Til Politisk Behandling – Skovveien
Oslo kommune Plan- og bygningsetaten Planforslag til politisk behandling – Skovveien Detaljregulering med konsekvensutredning Hensiktenmed planener å leggetil rette for at Briskebylinjenkan trafikkeresav nye trikker. Ruter AS foreslårå flytte Briskebytrikkenstrasé fra Inkognitogata/Riddervoldgatetil Skovveien (mellom Frognerveienog Briskebyveien)og å justeredagens trasé og holdeplasseri Briskebyveien.F orslaget vil bedrekollektivtrafikken s fremkommelighetog gi bedreforhold for gåendeog syklende. PBE anbefalerplanforslaget. Utarbeidetav: Norconsultfor Ruter AS Bydel: Frogner Saksnummer:201602328 Gnr./bnr.:213/437, 999/779, 999/282 m.fl. Dokumentnummer:121 Dato: 13.08.2018 Basertpå innsendtdoku m ent med dato: 25.05.2018 Husk alltid å oppgi saksnummer ved henvendelsetil Plan- og bygningsetaten. www.oslo.kommune.no/pbe Postadresse: Besøksadresse: Sentralbord, tlf: 21 80 21 80 [email protected] Boks 364 Sentrum Vahls gate 1 Kundesenteret, tlf: 23 49 10 00 0102 Oslo 0187 Oslo Bankgiro: 1315.01.01357 www.byplanoslo.no Org.nr.: NO 971 040 823 MVA Saksnr: 201602328-121 Side 2 av 123 Innhold 1 Hovedtrekk i planforslaget ............................................................................... 5 1.1 Sammendrag ......................................................................................................................... 5 1.1.1 Bakgrunn ..................................................................................................................... 5 1.1.2 Planforslaget .............................................................................................................. -

High-Level Forum Oslo 20/21 September 2010 UPDATE on the PRACTICAL INFORMATION
High-Level Forum Oslo 20/21 September 2010 HIGH-LEVEL FORUM ON JOBS FOR YOUTH OSLO, NORWAY 20-21 SEPTEMBER 2010 UPDATE ON THE PRACTICAL INFORMATION 1. Meeting Venue The Forum will be held at the Radisson Blu Scandinavia Hotel: http://www.radissonblu.com/scandinaviahotel-oslo on both days. Radisson Blu Scandinavia Hotel Holbergsgate 30, N-0166 Oslo, Norway Tel.: +47 23 29 30 00, Fax: +47 23 29 30 01 2. Bilateral meetings Bilateral meetings during the two days of the Forum will be possible, requests have to be addressed to Helena Treadwell-Guedj ([email protected], Tel: 01 45 24 92 51) by Thursday 16th September 3. Organisation of the meeting Please see the website: www.oecd.org/employment/youth/forum (English) www.oecd.org/emploi/jeunes/forum (French) Day One 20 September: Policy Forum (4 persons per Delegation) There will be interventions from different stakeholders: government, employer, union, youth organization representatives of governments, and social partners. Speakers wishing to make PowerPoint presentations during the Policy Forum are requested to send their presentations to the Secretariat by 16th September. They will also be made available on the website after the Forum. There are no registration fees. The lunch, offered by the Norwegian Ministry of Labour, will take place at the meeting venue in the Radisson Hotel. All participants are invited to attend a cocktail which will take place at 17.30 pm at the meeting venue in the Radisson Hotel. Day Two 21 September: Ministerial meeting (3 persons per Delegation) The second day is restricted to Ministers and other High-Level Officials. -

Annual Report 2004
Annual Report 2004 1 Contents Time for trains 3 What is Jernbaneverket? 4 Organisational structure 5 Safety 6 Finance and efficiency 10 Operations 10 Maintenance 11 Capital expenditure – rail network development 12 State Accounts for 2004 14 Human resources 16 Personnel and working environment 16 JBV Ressurs 16 Competitiveness 18 Train companies operating on the national rail network 18 Infrastructure capacity – Jernbaneverket’s core product 18 Operating parameters 19 Key figures for the national rail network 21 Traffic volumes on the national rail network 23 Punctuality 24 Environmental protection 26 International activities 28 Contact details 30 www.jernbaneverket.no 2 Cover: Jernbaneverket’s celebrations to mark 150 years of Norwegian railways. Photo: Øystein Grue Time for trains The past year marked the 150th anniversary of the railways in Norway and proved a worthy celebration. Punctuality has never been better, rail traffic is growing, and in summer 2004 the Norwegian Parliament took the historic decision to invest NOK 26.4 billion in developing a competitive rail network over the ten years from 2006 to 2015. In other words, the anniversary year not only provided the opportunity for a nostalgic look back, but also confirmed that the railways will continue to play a central role in the years ahead. In line with Parliament’s decision, value our good working relationship with autumn 2005. This brings us one step clo- Jernbaneverket has drawn up an action the trade unions. The railway has a culture ser to our goal of an efficient, modern rail programme which, if implemented, will and a historic legacy which need to be network in the Oslo region. -

20826 2011 Invitasjon.Indd
Invitation to the FIS Nordic World Ski Championships Holmenkollen, Oslo 2011 CONTENTS INVITATION .............................................................................................................................................................. 3 FACTS ABOUT OSLO ............................................................................................................................................ 4 ORGANIZATION ...................................................................................................................................................... 5 FIS OFFICIALS ......................................................................................................................................................... 6 PROGRAM ..................................................................................................................................................................7 FACTS ABOUT THE VENUES ............................................................................................................................ 8 Overview OF Courses ................................................................................................................................ 10 StadIUM MAPS .......................................................................................................................................................11 INFORMATION FROM THE ORGANIZER ................................................................................................... 15 FIS RULES ............................................................................................................................................................... -

Oslo Pass – the Official City Card See More
20 19 Oslo Pass – The Official City Card See more. Pay less Oslo Visitor Centre FROM: VALID ADULT 24 DATE MONTH YEAR TIME DATE _ / _/ : Exp: Dec 2021 • Free entry to 30 museums • Free public transport • Free entry to swimming pools • Discounts on sightseeing and special offers at restaurants, shops and leisure venues Pass type 24 hours 48 hours 72 hours Adult 445 NOK 655 NOK 820 NOK Child 235 NOK 325 NOK 410 NOK Senior 355 NOK 520 NOK 655 NOK CHILD: 6-17 YEARS, SENIOR: 67 YEARS + The 72h Oslo Pass incl. 24h free City Cruise (May-September). STUDENTS UP TO 30 YEARS OLD: 20% DISCOUNT Only offered at Oslo Visitor Centre and Ruter’s customer service office, and only by showing a valid student ID card with photo. Content How to use your Oslo Pass 04 The Mobile Oslo Pass 06 Contents of the Oslo Pass 08 Museums and attractions 14 Restaurants 46 Activities and special offers 56 Sightseeing 69 Conditions of use 77 Frequently asked questions 78 In 1984 the Oslo Pass was launched as one of the very first city cards in Europe. Thirty five years on, the Oslo Pass is still the best way to visit our city. The Oslo Pass includes free public transport and free entrance to museums and sights, in addition to discounts on activities and services. The Oslo Pass will save you time and money, and will allow you to discover the different parts of our beautiful city. You can purchase the Oslo Pass at the Oslo Visitor Centre inside Østbanehallen, by Oslo Central Station, where Oslo experts will help you plan your stay in our beautiful city. -
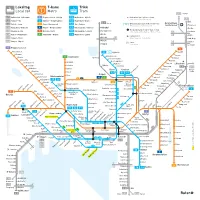
Lokaltog Local Rail Trikk Tram T-Bane Metro
Lokaltog T-bane Trikk Local rail Metro Tram L12 Eidsvoll L 1 Spikkestad – Lillestrøm 1 Frognerseteren – Helsfyr 11 Majorstuen – Kjelsås Holdeplass bare i pilens retning Stop in direction of arrow only L13 L 2 Skøyen – Ski 2 Gjønnes – Ellingsrudåsen 12 Majorstuen – Disen Dal L 3 Jaren Oslo lufthavn L 3 Oslo S – Jaren 3 Storo – Mortensrud 13 Jar – Grefsen 12 Endeholdeplass bare til bestemte tider Final stop at certain times only Gardermoen Hauerseter L12 Kongsberg – Eidsvoll 4 Ringen – Bergkrystallen 17 Rikshospitalet – Grefsen Hakadal Nordby Overgangsmuliget Tog / T-bane / Trikk Varingskollen L13 Drammen – Dal 5 Østerås – Vestli 18 Rikshospitalet – Holtet Interchange option Railway / Metro / Tram 4N Jessheim Åneby L14 Asker – Kongsvinger 6 Sognsvann – Ringen 19 Majorstuen – Ljabru Kløfta Flytogstasjon 3Ø Nittedal L21 Skøyen – Moss 2Ø Airport Express Train station Lindeberg Movatn 1 L22 Skøyen – Mysen Soner 3Ø Frogner Snippen 2V Fare zones 2Ø Leirsund 1 Frognerseteren 5 Voksenkollen 11 12 Kjelsås Vestli Lillevann Kjelsåsalleen Stovner Skogen 6 Sognsvann Kjelsås Grefsen stadion Rommen Voksenlia Grefsenplatået Romsås Kringsjå Holmenkollen Glads vei Grorud Lillestrøm Besserud Holstein Nydalen Sanatoriet Ammerud L 1 L14 Midtstuen Østhorn Disen Grefsen Kalbakken Sagdalen Kongs- Skådalen Tåsen Rødtvet vinger 12 13 17 Sinsenkrysset Strømmen Vettakollen Ringen Berg Veitvet Fjellhamar Gulleråsen Rikshospitalet Linderud 3 4 4 6 Hanaborg Gråkammen 17 18 Vollebekk Lørenskog Storo Sinsen Slemdal Nydalen 3 Risløkka Høybråten 2Ø Gaustad- Ullevål stadion -

Project Portfolio Infrastructure Construction Division
1 Project portfolio Infrastructure Construction Division www.banenor.no | 2019 3 We rely on close cooperation In the upcoming years there will be a large increase in About Bane NOR rail investments in Norway. As described in this folder the investments will be spread out on several projects. Bane NOR is a state-owned company respon- These investments create great value for all travelers. In the coming sible for the national railway infrastructure. Our years, departures will be more frequent, with reduced travel time mission is to ensure accessible railway infra- within the InterCity operating area. We are living in an exciting and Photo: Anne-Mette Storvik structure and efficient and user-friendly ser- changing infrastructure environment, with a high activity level. Over Einar Kilde vices, including the development of hubs and the next three years Bane NOR plans to introduce contracts relating goods terminals. to a large number of mega projects to the market. Investment will Executive Vice President continue until the InterCity rollout is completed as planned in 2034. Infrastructure Construction Division As of today, the Follo Line and Venjar-Langset are in production, The company’s main responsible are: and Sandvika-Moss-Såstad, Drammen-Kobbervikdalen and Ny- • Planning, development, administration, operation and kirke-Barkåker are out for tender. In 2019 and 2020 our plan is to maintenance of the national railway network • Traffic management start the pre-qualification process for Kleverud-Sørli-Åkersvika and • Administration and development of railway property The Ringerike Line and E16 highway. Additionally, Bane NOR plans together with The Norwegian Bane NOR has approximately 4,500 employees and the head office is Public Roads Administration, to build a safer and faster rail and road based in Oslo, Norway. -

On Track GLIMPSES of JERNBANEVERKET's ACTIVITIES in 2015
On track GLIMPSES OF JERNBANEVERKET'S ACTIVITIES IN 2015 Jernbaneverket is adopting new technology and new working methods in an increasing number of fields. A Norwegian railway tunnel is now being bored for the first time using TBM. Contact us Jernbaneverket units are located at several sites in the country. For more detailed information, visit our website or call our nationwide telephone service: 05280 From abroad (+47) 22 45 50 00 Postal address Jernbaneverket, Postboks 4350, NO-2308 Hamar Email [email protected] www.jernbaneverket.no “The major basic route change from December 2012 has resulted in formidable growth of passenger rail traffic in Eastern Norway, and the final pieces of the puzzle fell into place when Høvik station became fully operational by the time Contents of the timetable change in December 2015.” Editorial 3 Kjell Rune Pettersen Photo: Rail traffic 4 A glimmer of light for freight and rail services 4 12 CargoNet in the black 7 New tender 8 Timber on the increase 8 Punctuality approaching European peak 9 Maintenance and renewals 10 The beginning of a new era 11 Exciting times Firmly raising the standard at many stations 12 Rail initiative employed thousands 14 Hilde Lillejord Photo: A new era for tunnel building in Norway 16 In June 2015, the Parliament of Norway made to pave the way for further ERTMS development a decision to reform the railway sector. Work and digitalised infrastructure monitoring. Groundbreaking 18 relating to that reform has characterised the past year, and will do so to an even greater extent in In December, 17 km of new double track on the Ready for railway technology 18 Herrenknecht AG Photo: From concrete and ballast to steel and cables 19 2016. -

Velkommen Til Julemøte
VELKOMMEN TIL JULEMØTE Denne gang skal Sidsel Marie Nilsen snakke om sin far, Hans Jacob Nilsen, teatermann og idealist, på Persbråten videregående skole onsdag 24. november kl. 1800. Parkering på skolen. Enkel servering. Hans Jacob Nilsen som Peer Gynt på Det Norske Teatret. VELKOMMEN Finn Holden leder 1 BRUSTAD KOLONIAL PÅ SLEMDAL AV LAJLA HEYERDAHL Slemdal Sport er vel kjent. Etter nærmere undersøkelse viser det seg at eiendom men har en spennende og interessant historie. Frid Brustad er barnebarn av stifteren av Brustad Kolonial, Fredrik Wilhelm Brustad, født 1873. Da jeg ringte henne for å få noen opplysninger, måtte hun skuffe meg med at hun hadde kun et skjøte i en bankboks. Men da vi møttes en uke senere, hadde Frid fun net frem gamle album, bilder og brev datert 1898 fra kjelleren. Det er bevart histo riske dokumenter med håndskrevet skjøte og enestående fotografier som jeg gjerne vil dele med historielagets lesere. Bestefaren var den gang en forutseende, ung forretningsmann som startet sin første finere Colonial, Vin og Fedevarehandel i Bogstadveien 6 på hjørnet av Holtegaten. I sin reklame anbefaler han sine utsøkte varer fra de bestemte han seg for å avslutte butikken beste leverandører og fra egen import. og overdro rettigheten til Anders Larsen som startet utsalg på Lillebraaten. Etter hvert ble F. W. Brustad, som Lillebraaten hadde forretningen i Bogstadveien, Opprinnelig lå det et lite småbruk på interessert i å skaffe seg et nytt lokale haugen på Slemdal. Stedet het Lille til sin finere Colonial, Vin og Fedevare braaten og lå under Grimelund gård. I forretning. Brustad var på jakt etter 1898 solgte F.