Bacteriophages As Potential New Mammalian Pathogens George V
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
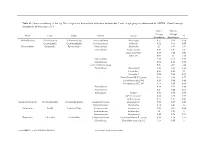
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention
J Neurogastroenterol Motil, Vol. 26 No. 4 October, 2020 pISSN: 2093-0879 eISSN: 2093-0887 https://doi.org/10.5056/jnm20080 JNM Journal of Neurogastroenterology and Motility Original Article Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention Choong-Kyun Noh and Kwang Jae Lee* Department of Gastroenterology, Ajou University School of Medicine, Suwon, Gyeonggi-do, Korea Background/Aims The pathophysiology of functional abdominal bloating and distention (FABD) is unclear yet. Our aim is to compare the diversity and composition of fecal microbiota in patients with FABD and healthy individuals, and to evaluate the relationship between small intestinal bacterial overgrowth (SIBO) and dysbiosis. Methods The microbiota of fecal samples was analyzed from 33 subjects, including 12 healthy controls and 21 patients with FABD diagnosed by the Rome IV criteria. FABD patients underwent a hydrogen breath test. Fecal microbiota composition was determined by 16S ribosomal RNA amplification and sequencing. Results Overall fecal microbiota composition of the FABD group differed from that of the control group. Microbial diversity was significantly lower in the FABD group than in the control group. Significantly higher proportion of Proteobacteria and significantly lower proportion of Actinobacteria were observed in FABD patients, compared with healthy controls. Compared with healthy controls, significantly higher proportion of Faecalibacterium in FABD patients and significantly higher proportion of Prevotella and Faecalibacterium in SIBO (+) patients with FABD were found. Faecalibacterium prausnitzii, was significantly more abundant, but Bacteroides uniformis and Bifidobacterium adolescentis were significantly less abundant in patients with FABD, compared with healthy controls. Significantly more abundant Prevotella copri and F. -

Metatranscriptomic Analysis to Define the Secrebiome, and 16S Rrna
Gallardo‑Becerra et al. Microb Cell Fact (2020) 19:61 https://doi.org/10.1186/s12934‑020‑01319‑y Microbial Cell Factories RESEARCH Open Access Metatranscriptomic analysis to defne the Secrebiome, and 16S rRNA profling of the gut microbiome in obesity and metabolic syndrome of Mexican children Luigui Gallardo‑Becerra1†, Fernanda Cornejo‑Granados1†, Rodrigo García‑López1†, Alejandra Valdez‑Lara1, Shirley Bikel1, Samuel Canizales‑Quinteros2, Blanca E. López‑Contreras2, Alfredo Mendoza‑Vargas3, Henrik Nielsen4 and Adrián Ochoa‑Leyva1* Abstract Background: In the last decade, increasing evidence has shown that changes in human gut microbiota are asso‑ ciated with diseases, such as obesity. The excreted/secreted proteins (secretome) of the gut microbiota afect the microbial composition, altering its colonization and persistence. Furthermore, it infuences microbiota‑host interac‑ tions by triggering infammatory reactions and modulating the host’s immune response. The metatranscriptome is essential to elucidate which genes are expressed under diseases. In this regard, little is known about the expressed secretome in the microbiome. Here, we use a metatranscriptomic approach to delineate the secretome of the gut microbiome of Mexican children with normal weight (NW) obesity (O) and obesity with metabolic syndrome (OMS). Additionally, we performed the 16S rRNA profling of the gut microbiota. Results: Out of the 115,712 metatranscriptome genes that codifed for proteins, 30,024 (26%) were predicted to be secreted, constituting the Secrebiome of the -

Depression and Microbiome—Study on the Relation and Contiguity Between Dogs and Humans
applied sciences Article Depression and Microbiome—Study on the Relation and Contiguity between Dogs and Humans Elisabetta Mondo 1,*, Alessandra De Cesare 1, Gerardo Manfreda 2, Claudia Sala 3 , Giuseppe Cascio 1, Pier Attilio Accorsi 1, Giovanna Marliani 1 and Massimo Cocchi 1 1 Department of Veterinary Medical Science, University of Bologna, Via Tolara di Sopra 50, 40064 Ozzano Emilia, Italy; [email protected] (A.D.C.); [email protected] (G.C.); [email protected] (P.A.A.); [email protected] (G.M.); [email protected] (M.C.) 2 Department of Agricultural and Food Sciences, University of Bologna, Via del Florio 2, 40064 Ozzano Emilia, Italy; [email protected] 3 Department of Physics and Astronomy, Alma Mater Studiorum, University of Bologna, 40126 Bologna, Italy; [email protected] * Correspondence: [email protected]; Tel.: +39-051-209-7329 Received: 22 November 2019; Accepted: 7 January 2020; Published: 13 January 2020 Abstract: Behavioral studies demonstrate that not only humans, but all other animals including dogs, can suffer from depression. A quantitative molecular evaluation of fatty acids in human and animal platelets has already evidenced similarities between people suffering from depression and German Shepherds, suggesting that domestication has led dogs to be similar to humans. In order to verify whether humans and dogs suffering from similar pathologies also share similar microorganisms at the intestinal level, in this study the gut-microbiota composition of 12 German Shepherds was compared to that of 15 dogs belonging to mixed breeds which do not suffer from depression. Moreover, the relation between the microbiota of the German Shepherd’s group and that of patients with depression has been investigated. -

Taxonomy of Lactobacilli and Bifidobacteria
Curr. Issues Intestinal Microbiol. 8: 44–61. Online journal at www.ciim.net Taxonomy of Lactobacilli and Bifdobacteria Giovanna E. Felis and Franco Dellaglio*† of carbohydrates. The genus Bifdobacterium, even Dipartimento Scientifco e Tecnologico, Facoltà di Scienze if traditionally listed among LAB, is only poorly MM. FF. NN., Università degli Studi di Verona, Strada le phylogenetically related to genuine LAB and its species Grazie 15, 37134 Verona, Italy use a metabolic pathway for the degradation of hexoses different from those described for ‘genuine’ LAB. Abstract The interest in what lactobacilli and bifdobacteria are Genera Lactobacillus and Bifdobacterium include a able to do must consider the investigation of who they large number of species and strains exhibiting important are. properties in an applied context, especially in the area of Before reviewing the taxonomy of those two food and probiotics. An updated list of species belonging genera, some basic terms and concepts and preliminary to those two genera, their phylogenetic relationships and considerations concerning bacterial systematics need to other relevant taxonomic information are reviewed in this be introduced: they are required for readers who are not paper. familiar with taxonomy to gain a deep understanding of The conventional nature of taxonomy is explained the diffculties in obtaining a clear taxonomic scheme for and some basic concepts and terms will be presented for the bacteria under analysis. readers not familiar with this important and fast-evolving area, -

Draft Genome and Description of Merdibacter Massiliensis Gen.Nov., Sp
www.nature.com/scientificreports OPEN Draft genome and description of Merdibacter massiliensis gen.nov., sp. nov., a new bacterium genus Received: 21 November 2018 Accepted: 9 May 2019 isolated from the human ileum Published: xx xx xxxx Hussein Anani1, Rita Abou Abdallah1, Nisrine Chelkha2, Anthony Fontanini2, Davide Ricaboni2, Morgane Mailhe2, Didier Raoult2,3 & Pierre-Edouard Fournier 1 We used phenotypic, genomic and phylogenetic information following the taxono-genomics approach to demonstrate that strain Marseille–P3254, isolated from an ileal sample of a 76-year old woman who underwent upper and lower digestive tract endoscopy for esophagitis and colonic polyp, is representative of a novel bacterial genus within the family Erysipelotrichaceae in the phylum Firmicutes. It is an anaerobic Gram-negative bacterium without catalase and oxidase activities. The genome of strain Marseille–P3254 is 2,468,496-bp long with a 40.1% G + C content. This new bacterium is most closely related to Eubacterium dolichum, with which it shares 90.7% 16S rRNA sequence similarity. In addition, genomic comparison using the digital DNA–DNA hybridization and OrthoANI analyses between the novel organism and the E. dolichum type strain revealed identities of 25.2 and 68.91%, respectively. The major fatty acids were C16: 0, C18: 1n9 and C18: 0. Based on these data, we propose the creation of the new genus Merdibacter gen. nov., with strain Marseille-P3254T (=CSUR P3254 = DSM 103534) being the type strain of the new species Merdibacter massiliensis gen. nov., sp. nov. A thorough knowledge of the gut microbiota composition appears essential to understand many aspects of health and diseases in humans. -

Ncomms4889.Pdf
ARTICLE Received 2 Jan 2014 | Accepted 14 Apr 2014 | Published 20 May 2014 DOI: 10.1038/ncomms4889 High-fat maternal diet during pregnancy persistently alters the offspring microbiome in a primate model Jun Ma1,2,*, Amanda L. Prince1,*, David Bader1,3,*, Min Hu1, Radhika Ganu1, Karalee Baquero4, Peter Blundell4, R. Alan Harris1,2, Antonio E. Frias4, Kevin L. Grove4 & Kjersti M. Aagaard1,2,3 The intestinal microbiome is a unique ecosystem and an essential mediator of metabolism and obesity in mammals. However, studies investigating the impact of the diet on the establishment of the gut microbiome early in life are generally lacking, and most notably so in primate models. Here we report that a high-fat maternal or postnatal diet, but not obesity per se, structures the offspring’s intestinal microbiome in Macaca fuscata (Japanese macaque). The resultant microbial dysbiosis is only partially corrected by a low-fat, control diet after weaning. Unexpectedly, early exposure to a high-fat diet diminished the abundance of non-pathogenic Campylobacter in the juvenile gut, suggesting a potential role for dietary fat in shaping commensal microbial communities in primates. Our data challenge the concept of an obesity-causing gut microbiome and rather provide evidence for a contribution of the maternal diet in establishing the microbiota, which in turn affects intestinal maintenance of metabolic health. 1 Departments of Obstetrics & Gynecology, Division of Maternal-Fetal Medicine at Baylor College of Medicine and Texas Children’s Hospital, Houston, Texas 77030, USA. 2 Department of Molecular and Human Genetics, Bioinformatics Research Lab at Baylor College of Medicine, Houston, Texas 77030, USA. -

The Dynamics of a Family's Gut Microbiota Reveal Variations on A
Schloss et al. Microbiome 2014, 2:25 http://www.microbiomejournal.com/content/2/1/25 RESEARCH Open Access The dynamics of a family’s gut microbiota reveal variations on a theme Patrick D Schloss1*, Kathryn D Iverson1, Joseph F Petrosino2 and Sarah J Schloss1 Abstract Background: It is clear that the structure and function of the human microbiota has significant impact on maintenance of health and yet the factors that give rise to an adult microbiota are poorly understood. A combination of genetics, diet, environment, and life history are all thought to impact the development of the gut microbiome. Here we study a chronosequence of the gut microbiota found in eight individuals from a family consisting of two parents and six children ranging in age from two months to ten years old. Results: Using 16S rRNA gene and metagenomic shotgun sequence data, it was possible to distinguish the family from a cohort of normal individuals living in the same geographic region and to differentiate each family member. Interestingly, there was a significant core membership to the family members’ microbiota where the abundance of this core accounted for the differences between individuals. It was clear that the introduction of solids represents a significant transition in the development of a mature microbiota. This transition was associated with increased diversity, decreased stability, and the colonization of significant abundances of Bacteroidetes and Clostridiales. Although the children and mother shared essentially the identical diet and environment, the children’s microbiotas were not significantly more similar to their mother than they were to their father. Conclusions: This analysis underscores the complex interactions that give rise to a personalized microbiota and suggests the value of studying families as a surrogate for longitudinal studies. -

The Gut Microbiome in Dogs with Congestive Heart Failure: a Pilot Study Joonbum Seo1*, Linda Matthewman2, Dong Xia3, Jenny Wilshaw1, Yu‑Mei Chang3 & David J
www.nature.com/scientificreports OPEN The gut microbiome in dogs with congestive heart failure: a pilot study Joonbum Seo1*, Linda Matthewman2, Dong Xia3, Jenny Wilshaw1, Yu‑Mei Chang3 & David J. Connolly1 Compromised gut health and dysbiosis in people with heart failure has received a great deal of attention over the last decade. Whether dogs with heart failure have a similar dysbiosis pattern to what is described in people is currently unknown. We hypothesised that dogs with congestive heart failure have quantifable dysbiosis compared to healthy dogs that are similar in sex and age. A total of 50 dogs (15 healthy dogs and 35 dogs with congestive heart failure) were prospectively recruited, and their faecal gut microbiome was assessed using 16S rRNA sequencing (Illumina MiSeq platform). There was no signifcant change in the microbial diversity and richness in dogs with congestive heart failure. However, there was an increase in abundance of Proteobacteria in the congestive heart failure group (p = 0.014), particularly due to an increase in the family Enterobacteriaceae (p = 0.002) and Escherichia coli (p = 0.033). We conclude that dogs with congestive heart failure have dysbiosis, and we show additional trends in our data suggesting that dogs may have a similar pattern to that described in people. The results of this study provide useful preliminary information and raise the possibility that dogs represent a clinically relevant animal model of dysbiosis in people with heart failure. Te gut microbiota is a consortium of microorganisms in the digestive tract 1. In addition to contributing to digestion, these organisms function as an endocrine organ through the production of bioactive metabolites and modulate the immune system through the translocation of immunomodulatory bacterial products across the intestinal wall1. -

Diversity of the Human Gastrointestinal Microbiota Novel Perspectives from High Throughput Analyses
Diversity of the Human Gastrointestinal Microbiota Novel Perspectives from High Throughput Analyses Mirjana Rajilić-Stojanović Promotor Prof. Dr W. M. de Vos Hoogleraar Microbiologie Wageningen Universiteit Samenstelling Promotiecommissie Prof. Dr T. Abee Wageningen Universiteit Dr J. Doré INRA, France Prof. Dr G. R. Gibson University of Reading, UK Prof. Dr S. Salminen University of Turku, Finland Dr K. Venema TNO Quality for life, Zeist Dit onderzoek is uitgevoerd binnen de onderzoekschool VLAG. Diversity of the Human Gastrointestinal Microbiota Novel Perspectives from High Throughput Analyses Mirjana Rajilić-Stojanović Proefschrift Ter verkrijging van de graad van doctor op gezag van de rector magnificus van Wageningen Universiteit, Prof. dr. M. J. Kropff, in het openbaar te verdedigen op maandag 11 juni 2007 des namiddags te vier uur in de Aula Mirjana Rajilić-Stojanović, Diversity of the Human Gastrointestinal Microbiota – Novel Perspectives from High Throughput Analyses (2007) PhD Thesis, Wageningen University and Research Centre, Wageningen, The Netherlands – with Summary in Dutch – 214 p. ISBN: 978-90-8504-663-9 Abstract The human gastrointestinal tract is densely populated by hundreds of microbial (primarily bacterial, but also archaeal and fungal) species that are collectively named the microbiota. This microbiota performs various functions and contributes significantly to the digestion and the health of the host. It has previously been noted that the diversity of the gastrointestinal microbiota is disturbed in relation to several intestinal and not intestine related diseases. However, accurate and detailed defining of such disturbances is hampered by the fact that the diversity of this ecosystem is still not fully described, primarily because of its extreme complexity, and high inter-individual variability. -

Enlightening the Taxonomy Darkness of Human Gut Microbiomes with a Cultured Biobank
Enlightening the Taxonomy Darkness of Human Gut Microbiomes With a Cultured Biobank Chang Liu Institute of Microbiology Chinese Academy of Sciences Mengxuan Du Institute of Microbiology Chinese Academy of Sciences Rexiding Abuduaini Institute of Microbiology Chinese Academy of Sciences Hai-Ying Yu Institute of Microbiology Chinese Academy of Sciences Dan-Hua Li Institute of Microbiology Chinese Academy of Sciences Yu-Jing Wang Institute of Microbiology Chinese Academy of Sciences Nan Zhou Institute of Microbiology Chinese Academy of Sciences Min-Zhi Jiang Institute of Microbiology Chinese Academy of Sciences Peng-Xia Niu Institute of Microbiology Chinese Academy of Sciences Shan-Shan Han Institute of Microbiology Chinese Academy of Sciences Hong-He Chen Institute of Microbiology Chinese Academy of Sciences Wen-Yu Shi Institute of Microbiology Chinese Academy of Sciences Linhuan Wu Institute of Microbiology Chinese Academy of Sciences Yu-Hua Xin Institute of Microbiology Chinese Academy of Sciences Juncai Ma Institute of Microbiology Chinese Academy of Sciences Yuguang Zhou Institute of Microbiology Chinese Academy of Sciences Cheng-Ying Jiang Institute of Microbiology Chinese Academy of Sciences Hong-wei Liu Page 1/52 Institute of Microbiology Chinese Academy of Sciences Shuang-Jiang Liu ( [email protected] ) Institute of Microbiology Chinese Academy of Sciences https://orcid.org/0000-0002-7585-310X Research Keywords: human gut microbiomes, cultivation, biobank, novel taxa, hGMB Posted Date: February 8th, 2021 DOI: https://doi.org/10.21203/rs.3.rs-168279/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 2/52 Abstract Background The cultivated gut microbial resource plays essential roles in gut microbiome studies such as unraveling gut microbial functions and host-microbe interactions. -

Dose-Dependent Effects of Dietary Xylooligosaccharides
molecules Article Dose-Dependent Effects of Dietary Xylooligosaccharides Supplementation on Microbiota, Fermentation and Metabolism in Healthy Adult Cats 1, 1, 2 2 1 Yang Lyu y , Sandra Debevere y , Hermann Bourgeois , Mavis Ran , Bart J.G. Broeckx , Lynn Vanhaecke 3, Tom Van de Wiele 4 and Myriam Hesta 2,* 1 Department of Nutrition, Genetics and Ethology, Faculty of Veterinary Medicine, Ghent University, Heidestraat 19, 9820 Merelbeke, Belgium; [email protected] (Y.L.); [email protected] (S.D.); [email protected] (B.J.G.B.) 2 Longlive Biotechnology Co. Ltd., Haier Road 63, Qingdao 266103, China; [email protected] (H.B.); [email protected] (M.R.) 3 Lab of Chemical Analysis, Faculty of Veterinary Medicine, Ghent University, Salisburylaan 133, 9820 Merelbeke, Belgium; [email protected] 4 Center for Microbial Ecology and Technology (CMET), Faculty of Bioscience Engineering, Ghent University, Coupure Links 653, 9000 Ghent, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +32(0)92647827 These authors contributed equally to this work. y Academic Editor: Solomon Habtemariam Received: 6 October 2020; Accepted: 27 October 2020; Published: 29 October 2020 Abstract: In order to investigate the effect and appropriate dose of prebiotics, this study evaluated the effect of two levels of xylooligosaccharides (XOS) in cats. Twenty-four healthy adult cats were divided into three groups: no-XOS control diet with 1% cellulose; low XOS supplementation (LXOS) with 0.04% XOS and 0.96% cellulose; and high XOS supplementation (HXOS) with 0.40% XOS and 0.60% cellulose. Both XOS groups increased blood 3-hydroxybutyryl carnitine levels and decreased hexadecanedioyl carnitine levels.