1,520 Reference Genomes from Cultivated Human Gut Bacteria Enable Functional Microbiome Analyses
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

A Taxonomic Note on the Genus Lactobacillus
Taxonomic Description template 1 A taxonomic note on the genus Lactobacillus: 2 Description of 23 novel genera, emended description 3 of the genus Lactobacillus Beijerinck 1901, and union 4 of Lactobacillaceae and Leuconostocaceae 5 Jinshui Zheng1, $, Stijn Wittouck2, $, Elisa Salvetti3, $, Charles M.A.P. Franz4, Hugh M.B. Harris5, Paola 6 Mattarelli6, Paul W. O’Toole5, Bruno Pot7, Peter Vandamme8, Jens Walter9, 10, Koichi Watanabe11, 12, 7 Sander Wuyts2, Giovanna E. Felis3, #*, Michael G. Gänzle9, 13#*, Sarah Lebeer2 # 8 '© [Jinshui Zheng, Stijn Wittouck, Elisa Salvetti, Charles M.A.P. Franz, Hugh M.B. Harris, Paola 9 Mattarelli, Paul W. O’Toole, Bruno Pot, Peter Vandamme, Jens Walter, Koichi Watanabe, Sander 10 Wuyts, Giovanna E. Felis, Michael G. Gänzle, Sarah Lebeer]. 11 The definitive peer reviewed, edited version of this article is published in International Journal of 12 Systematic and Evolutionary Microbiology, https://doi.org/10.1099/ijsem.0.004107 13 1Huazhong Agricultural University, State Key Laboratory of Agricultural Microbiology, Hubei Key 14 Laboratory of Agricultural Bioinformatics, Wuhan, Hubei, P.R. China. 15 2Research Group Environmental Ecology and Applied Microbiology, Department of Bioscience 16 Engineering, University of Antwerp, Antwerp, Belgium 17 3 Dept. of Biotechnology, University of Verona, Verona, Italy 18 4 Max Rubner‐Institut, Department of Microbiology and Biotechnology, Kiel, Germany 19 5 School of Microbiology & APC Microbiome Ireland, University College Cork, Co. Cork, Ireland 20 6 University of Bologna, Dept. of Agricultural and Food Sciences, Bologna, Italy 21 7 Research Group of Industrial Microbiology and Food Biotechnology (IMDO), Vrije Universiteit 22 Brussel, Brussels, Belgium 23 8 Laboratory of Microbiology, Department of Biochemistry and Microbiology, Ghent University, Ghent, 24 Belgium 25 9 Department of Agricultural, Food & Nutritional Science, University of Alberta, Edmonton, Canada 26 10 Department of Biological Sciences, University of Alberta, Edmonton, Canada 27 11 National Taiwan University, Dept. -

Macellibacteroides Fermentans Gen. Nov., Sp. Nov., a Member of the Family Porphyromonadaceae Isolated from an Upflow Anaerobic Filter Treating Abattoir Wastewaters
International Journal of Systematic and Evolutionary Microbiology (2012), 62, 2522–2527 DOI 10.1099/ijs.0.032508-0 Macellibacteroides fermentans gen. nov., sp. nov., a member of the family Porphyromonadaceae isolated from an upflow anaerobic filter treating abattoir wastewaters Linda Jabari,1,2 Hana Gannoun,2 Jean-Luc Cayol,1 Abdeljabbar Hedi,1 Mitsuo Sakamoto,3 Enevold Falsen,4 Moriya Ohkuma,3 Moktar Hamdi,2 Guy Fauque,1 Bernard Ollivier1 and Marie-Laure Fardeau1 Correspondence 1Aix-Marseille Universite´ du Sud Toulon-Var, CNRS/INSU, IRD, MIO, UM 110, Case 925, Marie-Laure Fardeau 163 Avenue de Luminy, 13288 Marseille Cedex 9, France [email protected] 2Laboratoire d’Ecologie et de Technologie Microbienne, Institut National des Sciences Applique´es et de Technologie, Centre Urbain Nord, BP 676, 1080 Tunis Cedex, Tunisia 3Microbe Division/Japan Collection of Microorganisms, RIKEN BioResource Center 2-1 Hirosawa, Wako, Saitama 351-0198, Japan 4CCUG, Culture Collection, Department of Clinical Bacteriology, University of Go¨teborg, 41346 Go¨teborg, Sweden A novel obligately anaerobic, non-spore-forming, rod-shaped mesophilic bacterium, which stained Gram-positive but showed the typical cell wall structure of Gram-negative bacteria, was isolated from an upflow anaerobic filter treating abattoir wastewaters in Tunisia. The strain, designated LIND7HT, grew at 20–45 6C (optimum 35–40 6C) and at pH 5.0–8.5 (optimum pH 6.5–7.5). It did not require NaCl for growth, but was able to grow in the presence of up to 2 % NaCl. Sulfate, thiosulfate, elemental sulfur, sulfite, nitrate and nitrite were not used as terminal electron acceptors. -

Robust Taxonomic Classification of Uncharted Microbial Sequences and Bins with CAT and BAT
bioRxiv preprint doi: https://doi.org/10.1101/530188; this version posted January 24, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT F.A. Bastiaan von Meijenfeldt1,†, Ksenia Arkhipova1,†, Diego D. Cambuy1, Felipe H. Coutinho2,3, Bas E. Dutilh1,2,* 1 Theoretical Biology and Bioinformatics, Science for Life, Utrecht University, The Netherlands. 2 Centre for Molecular and Biomolecular Informatics, Radboud University Medical Centre, Nijmegen, The Netherlands. 3 Instituto de Biologia, Universidade Federal do Rio de Janeiro, Rio de Janeiro, RJ, Brazil. * To whom correspondence should be addressed. Tel: +31 30 253 4212; Email: [email protected]. † These authors contributed equally to this work. Present Address: [Felipe H. Couthinho], Evolutionary Genomics Group, Departamento de Produccíon y Microbiología, Universidad Miguel Hernández, Campus San Juan, San Juan, Alicante 03550, Spain. ABSTRACT Current-day metagenomics increasingly requires taxonomic classification of long DNA sequences and metagenome-assembled genomes (MAGs) of unknown microorganisms. We show that the standard best-hit approach often leads to classifications that are too specific. We present tools to classify high- quality metagenomic contigs (Contig Annotation Tool, CAT) and MAGs (Bin Annotation Tool, BAT) and thoroughly benchmark them with simulated metagenomic sequences that are classified against a reference database where related sequences are increasingly removed, thereby simulating increasingly unknown queries. We find that the query sequences are correctly classified at low taxonomic ranks if closely related organisms are present in the reference database, while classifications are made higher in the taxonomy when closely related organisms are absent, thus avoiding spurious classification specificity. -

Gut Microbiota and Inflammation
Nutrients 2011, 3, 637-682; doi:10.3390/nu3060637 OPEN ACCESS nutrients ISSN 2072-6643 www.mdpi.com/journal/nutrients Review Gut Microbiota and Inflammation Asa Hakansson and Goran Molin * Food Hygiene, Division of Applied Nutrition, Department of Food Technology, Engineering and Nutrition, Lund University, PO Box 124, SE-22100 Lund, Sweden; E-Mail: [email protected] * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +46-46-222-8327; Fax: +46-46-222-4532. Received: 15 April 2011; in revised form: 19 May 2011 / Accepted: 24 May 2011 / Published: 3 June 2011 Abstract: Systemic and local inflammation in relation to the resident microbiota of the human gastro-intestinal (GI) tract and administration of probiotics are the main themes of the present review. The dominating taxa of the human GI tract and their potential for aggravating or suppressing inflammation are described. The review focuses on human trials with probiotics and does not include in vitro studies and animal experimental models. The applications of probiotics considered are systemic immune-modulation, the metabolic syndrome, liver injury, inflammatory bowel disease, colorectal cancer and radiation-induced enteritis. When the major genomic differences between different types of probiotics are taken into account, it is to be expected that the human body can respond differently to the different species and strains of probiotics. This fact is often neglected in discussions of the outcome of clinical trials with probiotics. Keywords: probiotics; inflammation; gut microbiota 1. Inflammation Inflammation is a defence reaction of the body against injury. The word inflammation originates from the Latin word ―inflammatio‖ which means fire, and traditionally inflammation is characterised by redness, swelling, pain, heat and impaired body functions. -

Integrative Annotation of 21,037 Human Genes Validated by Full-Length Cdna Clones
PLoS BIOLOGY Integrative Annotation of 21,037 Human Genes Validated by Full-Length cDNA Clones Tadashi Imanishi1, Takeshi Itoh1,2, Yutaka Suzuki3,68, Claire O’Donovan4, Satoshi Fukuchi5, Kanako O. Koyanagi6, Roberto A. Barrero5, Takuro Tamura7,8, Yumi Yamaguchi-Kabata1, Motohiko Tanino1,7, Kei Yura9, Satoru Miyazaki5, Kazuho Ikeo5, Keiichi Homma5, Arek Kasprzyk4, Tetsuo Nishikawa10,11, Mika Hirakawa12, Jean Thierry-Mieg13,14, Danielle Thierry-Mieg13,14, Jennifer Ashurst15, Libin Jia16, Mitsuteru Nakao3, Michael A. Thomas17, Nicola Mulder4, Youla Karavidopoulou4, Lihua Jin5, Sangsoo Kim18, Tomohiro Yasuda11, Boris Lenhard19, Eric Eveno20,21, Yoshiyuki Suzuki5, Chisato Yamasaki1, Jun-ichi Takeda1, Craig Gough1,7, Phillip Hilton1,7, Yasuyuki Fujii1,7, Hiroaki Sakai1,7,22, Susumu Tanaka1,7, Clara Amid23, Matthew Bellgard24, Maria de Fatima Bonaldo25, Hidemasa Bono26, Susan K. Bromberg27, Anthony J. Brookes19, Elspeth Bruford28, Piero Carninci29, Claude Chelala20, Christine Couillault20,21, Sandro J. de Souza30, Marie-Anne Debily20, Marie-Dominique Devignes31, Inna Dubchak32, Toshinori Endo33, Anne Estreicher34, Eduardo Eyras15, Kaoru Fukami-Kobayashi35, Gopal R. Gopinath36, Esther Graudens20,21, Yoonsoo Hahn18, Michael Han23, Ze-Guang Han21,37, Kousuke Hanada5, Hideki Hanaoka1, Erimi Harada1,7, Katsuyuki Hashimoto38, Ursula Hinz34, Momoki Hirai39, Teruyoshi Hishiki40, Ian Hopkinson41,42, Sandrine Imbeaud20,21, Hidetoshi Inoko1,7,43, Alexander Kanapin4, Yayoi Kaneko1,7, Takeya Kasukawa26, Janet Kelso44, Paul Kersey4, Reiko Kikuno45, Kouichi -

Gut Microbiota Features Associated with Clostridioides Difficile Colonization in Dairy Calves
bioRxiv preprint doi: https://doi.org/10.1101/2021.05.11.443551; this version posted May 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Gut microbiota features associated with Clostridioides 2 difficile colonization in dairy calves 3 4 Laurel E. Redding1, Alexander S. Berry2,3, Nagaraju Indugu1, Elizabeth Huang1, Daniel P. Beiting3, Dipti 5 Pitta1 6 7 8 1Department of Clinical Sciences, University of Pennsylvania, School of Veterinary Medicine, Kennett 9 Square, PA, USA 10 11 2Division of Gastroenterology, Hepatology, and Nutrition, Children’s Hospital of Philadelphia, Philadelphia, 12 PA, USA 13 14 3Department of Pathobiology, School of Veterinary Medicine, University of Pennsylvania, Philadelphia, PA, 15 USA 16 17 18 Corresponding author: Laurel Redding, [email protected] 19 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.05.11.443551; this version posted May 11, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 20 Abstract 21 Diarrheal disease, a major cause of morbidity and mortality in dairy calves, is strongly associated with the 22 health and composition of the gut microbiome. Clostridioides difficile is an opportunistic pathogen that 23 proliferates and can produce enterotoxins when the host experiences gut dysbiosis. -
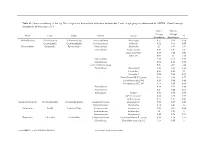
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Acidaminococcus Provencensis Sp. Nov., a New Bacterium Isolated from a Fresh Human Stool Specimen
NEW SPECIES Acidaminococcus provencensis sp. nov., a new bacterium isolated from a fresh human stool specimen T. Takakura1,2, H. Anani3, A. Fadlane2, A. Fontanini2, D. Raoult2,3 and J. Y. Bou Khalil2 1) Hitachi High-Technologies Corporation, Analytical & Medical Solution Business Group, Ibaraki-ken, Japan, 2) Institut Hospitalo-Universitaire Méditerranée- Infection, Marseille, France and 3) Aix-Marseille Université, Institut de Recherche pour le Developpement (IRD), UMR Microbes Evolution Phylogeny and Infections (MEPHI), Marseille, France Abstract Acidaminococcus provencensis strain Marseille-P4266T (= CSURP4266T) is a new species isolated from a fresh human stool specimen. © 2019 The Author(s). Published by Elsevier Ltd. Keywords: Acidaminococcus provencensis, Culturomics, Microbiota, New species, Taxonogenomics, TM4000, Stool Original Submission: 17 May 2019; Accepted: 5 June 2019 Article published online: 14 June 2019 imported into MALDI BIOTYPER 3.0 software (Bruker Dal- Corresponding author: J.Y. Bou Khalil, MEPHI, Institut Hospitalo- tonics) and analysed against the main spectra of the bacteria Universitaire Méditerranée Infection, 19–21 Boulevard Jean Moulin, 13005, Marseille Cedex 05, France. included in two databases (Bruker and the constantly updated E-mail: [email protected] MEPHI databases: https://www.mediterranee-infection.com/ urms-data-base/). The study was validated by the ethics com- mittee of the Institut Fédératif de Recherche IFR48 under number 09-022. Initial growth was obtained after 72 h of Introduction culture in Colombia agar enriched with 5% sheep’s blood (bioMérieux, Marcy l’Etoile, France) in strict anaerobic con- ditions at 37°C. Culturomics is a concept that involves the development of different culture conditions to enlarge our knowledge of the human microbiota through the discovery of previously uncul- Phenotypic characteristics tured bacteria [1–4]. -
![Download Or Browsing at Phylomedb [118]](https://docslib.b-cdn.net/cover/5001/download-or-browsing-at-phylomedb-118-835001.webp)
Download Or Browsing at Phylomedb [118]
Gerdol et al. Genome Biology (2020) 21:275 https://doi.org/10.1186/s13059-020-02180-3 RESEARCH Open Access Massive gene presence-absence variation shapes an open pan-genome in the Mediterranean mussel Marco Gerdol1, Rebeca Moreira2, Fernando Cruz3, Jessica Gómez-Garrido3, Anna Vlasova4, Umberto Rosani5, Paola Venier5, Miguel A. Naranjo-Ortiz4,6, Maria Murgarella7, Samuele Greco1, Pablo Balseiro2,8, André Corvelo3,9, Leonor Frias3, Marta Gut3, Toni Gabaldón4,6,10,11, Alberto Pallavicini1,12, Carlos Canchaya7,13,14, Beatriz Novoa2, Tyler S. Alioto3,6, David Posada7,13,14* and Antonio Figueras2* * Correspondence: dposada@uvigo. es; [email protected] Abstract 7Department of Biochemistry, Genetics and Immunology, Background: The Mediterranean mussel Mytilus galloprovincialis is an ecologically University of Vigo, 36310 Vigo, and economically relevant edible marine bivalve, highly invasive and resilient to Spain biotic and abiotic stressors causing recurrent massive mortalities in other bivalves. 2Instituto de Investigaciones Marinas (IIM - CSIC), Eduardo Although these traits have been recently linked with the maintenance of a high Cabello, 6, 36208 Vigo, Spain genetic variation within natural populations, the factors underlying the evolutionary Full list of author information is success of this species remain unclear. available at the end of the article Results: Here, after the assembly of a 1.28-Gb reference genome and the resequencing of 14 individuals from two independent populations, we reveal a complex pan-genomic architecture in M. galloprovincialis, with a core set of 45,000 genes plus a strikingly high number of dispensable genes (20,000) subject to presence-absence variation, which may be entirely missing in several individuals. -

Description of Gabonibacter Massiliensis Gen. Nov., Sp. Nov., a New Member of the Family Porphyromonadaceae Isolated from the Human Gut Microbiota
Curr Microbiol DOI 10.1007/s00284-016-1137-2 Description of Gabonibacter massiliensis gen. nov., sp. nov., a New Member of the Family Porphyromonadaceae Isolated from the Human Gut Microbiota 1,2 1 3,4 Gae¨l Mourembou • Jaishriram Rathored • Jean Bernard Lekana-Douki • 5 1 1 Ange´lique Ndjoyi-Mbiguino • Saber Khelaifia • Catherine Robert • 1 1,6 1 Nicholas Armstrong • Didier Raoult • Pierre-Edouard Fournier Received: 9 June 2016 / Accepted: 8 September 2016 Ó Springer Science+Business Media New York 2016 Abstract The identification of human-associated bacteria Gabonibacter gen. nov. and the new species G. mas- is very important to control infectious diseases. In recent siliensis gen. nov., sp. nov. years, we diversified culture conditions in a strategy named culturomics, and isolated more than 100 new bacterial Keywords Gabonibacter massiliensis Á Taxonogenomics Á species and/or genera. Using this strategy, strain GM7, a Culturomics Á Gabon Á Gut microbiota strictly anaerobic gram-negative bacterium was recently isolated from a stool specimen of a healthy Gabonese Abbreviations patient. It is a motile coccobacillus without catalase and CSUR Collection de Souches de l’Unite´ des oxidase activities. The genome of Gabonibacter mas- Rickettsies siliensis is 3,397,022 bp long with 2880 ORFs and a G?C DSM Deutsche Sammlung von content of 42.09 %. Of the predicted genes, 2,819 are Mikroorganismen protein-coding genes, and 61 are RNAs. Strain GM7 differs MALDI-TOF Matrix-assisted laser desorption/ from the closest genera within the family Porphyromon- MS ionization time-of-flight mass adaceae both genotypically and in shape and motility. -

Alterations in the Gut Bacterial Microbiome in Fungal Keratitis Patients
RESEARCH ARTICLE Alterations in the gut bacterial microbiome in fungal Keratitis patients Sama Kalyana Chakravarthy1☯, Rajagopalaboopathi Jayasudha1☯, Konduri Ranjith1, Anirban Dutta2, Nishal Kumar Pinna2, Sharmila S. Mande2, Savitri Sharma1, Prashant Garg3, Somasheila I. Murthy3, Sisinthy Shivaji1* 1 Jhaveri Microbiology Centre, Brien Holden Eye Research Centre, L. V. Prasad Eye Institute, Kallam Anji Reddy campus, Hyderabad, India, 2 Bio-Sciences R&D Division, TCS Research, Tata Consultancy Services Ltd., Pune, India, 3 Tej Kohli Cornea Institute, L. V. Prasad Eye Institute, Kallam Anji Reddy campus, Hyderabad, India a1111111111 a1111111111 ☯ These authors contributed equally to this work. a1111111111 * [email protected] a1111111111 a1111111111 Abstract Dysbiosis in the gut microbiome has been implicated in several diseases including auto- immune diseases, inflammatory diseases, cancers and mental disorders. Keratitis is an OPEN ACCESS inflammatory disease of the eye significantly contributing to corneal blindness in the devel- Citation: Kalyana Chakravarthy S, Jayasudha R, oping world. It would be worthwhile to investigate the possibility of dysbiosis in the gut micro- Ranjith K, Dutta A, Pinna NK, Mande SS, et al. (2018) Alterations in the gut bacterial microbiome biome being associated with Keratitis. Here, we have analyzed fungal and bacterial in fungal Keratitis patients. PLoS ONE 13(6): populations in stool samples through high-throughput sequencing of the ITS2 region for e0199640. https://doi.org/10.1371/journal. fungi and V3-V4 region of 16S rRNA gene for bacteria in healthy controls (HC, n = 31) and pone.0199640 patients with fungal keratitis (FK, n = 32). Candida albicans (2 OTUs), Aspergillus (1 OTU) Editor: Suresh Yenugu, University of Hyderabad, and 3 other denovo-OTUs were enriched in FK samples and an unclassified denovo-OTU INDIA was enriched in HC samples. -

Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention
J Neurogastroenterol Motil, Vol. 26 No. 4 October, 2020 pISSN: 2093-0879 eISSN: 2093-0887 https://doi.org/10.5056/jnm20080 JNM Journal of Neurogastroenterology and Motility Original Article Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention Choong-Kyun Noh and Kwang Jae Lee* Department of Gastroenterology, Ajou University School of Medicine, Suwon, Gyeonggi-do, Korea Background/Aims The pathophysiology of functional abdominal bloating and distention (FABD) is unclear yet. Our aim is to compare the diversity and composition of fecal microbiota in patients with FABD and healthy individuals, and to evaluate the relationship between small intestinal bacterial overgrowth (SIBO) and dysbiosis. Methods The microbiota of fecal samples was analyzed from 33 subjects, including 12 healthy controls and 21 patients with FABD diagnosed by the Rome IV criteria. FABD patients underwent a hydrogen breath test. Fecal microbiota composition was determined by 16S ribosomal RNA amplification and sequencing. Results Overall fecal microbiota composition of the FABD group differed from that of the control group. Microbial diversity was significantly lower in the FABD group than in the control group. Significantly higher proportion of Proteobacteria and significantly lower proportion of Actinobacteria were observed in FABD patients, compared with healthy controls. Compared with healthy controls, significantly higher proportion of Faecalibacterium in FABD patients and significantly higher proportion of Prevotella and Faecalibacterium in SIBO (+) patients with FABD were found. Faecalibacterium prausnitzii, was significantly more abundant, but Bacteroides uniformis and Bifidobacterium adolescentis were significantly less abundant in patients with FABD, compared with healthy controls. Significantly more abundant Prevotella copri and F.