Metatranscriptomic Analysis to Define the Secrebiome, and 16S Rrna
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Human Gut Microbiome Changes During a 10 Week Randomised
www.nature.com/scientificreports Corrected: Author Correction OPEN Human gut microbiome changes during a 10 week Randomised Control Trial for micronutrient Received: 19 July 2018 Accepted: 20 June 2019 supplementation in children with Published online: 12 July 2019 attention defcit hyperactivity disorder Aaron J. Stevens1, Rachel V. Purcell 2, Kathryn A. Darling3, Matthew J. F. Eggleston4, Martin A. Kennedy 1 & Julia J. Rucklidge3 It has been widely hypothesized that both diet and the microbiome play a role in the regulation of attention-defcit/hyperactivity disorder (ADHD) behaviour. However, there has been very limited scientifc investigation into the potential biological connection. We performed a 10-week pilot study investigating the efects of a broad spectrum micronutrient administration on faecal microbiome content, using 16S rRNA gene sequencing. The study consisted of 17 children (seven in the placebo and ten in the treatment group) between the ages of seven and 12 years, who were diagnosed with ADHD. We found that micronutrient treatment did not drive large-scale changes in composition or structure of the microbiome. However, observed OTUs signifcantly increased in the treatment group, and showed no mean change in the placebo group. The diferential abundance and relative frequency of Actinobacteria signifcantly decreased post- micronutrient treatment, and this was largely attributed to species from the genus Bifdobacterium. This was compensated by an increase in the relative frequency of species from the genus Collinsella. Further research is required to establish the role that Bifdobacterium contribute towards neuropsychiatric disorders; however, these fndings suggest that micronutrient administration could be used as a safe, therapeutic method to modulate Bifdobacterium abundance, which could have potential implications for modulating and regulating ADHD behaviour. -

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -
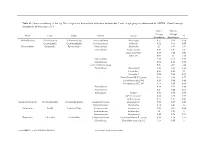
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Early-Life Gut Dysbiosis Linked to Juvenile Mortality in Ostriches Elin Videvall1,2* , Se Jin Song3,4, Hanna M
Videvall et al. Microbiome (2020) 8:147 https://doi.org/10.1186/s40168-020-00925-7 RESEARCH Open Access Early-life gut dysbiosis linked to juvenile mortality in ostriches Elin Videvall1,2* , Se Jin Song3,4, Hanna M. Bensch1, Maria Strandh1, Anel Engelbrecht5, Naomi Serfontein6, Olof Hellgren1, Adriaan Olivier7, Schalk Cloete5,8, Rob Knight3,4,9,10 and Charlie K. Cornwallis1 Abstract Background: Imbalances in the gut microbial community (dysbiosis) of vertebrates have been associated with several gastrointestinal and autoimmune diseases. However, it is unclear which taxa are associated with gut dysbiosis, and if particular gut regions or specific time periods during ontogeny are more susceptible. We also know very little of this process in non-model organisms, despite an increasing realization of the general importance of gut microbiota for health. Methods: Here, we examine the changes that occur in the microbiome during dysbiosis in different parts of the gastrointestinal tract in a long-lived bird with high juvenile mortality, the ostrich (Struthio camelus). We evaluated the 16S rRNA gene composition of the ileum, cecum, and colon of 68 individuals that died of suspected enterocolitis during the first 3 months of life (diseased individuals), and of 50 healthy individuals that were euthanized as age-matched controls. We combined these data with longitudinal environmental and fecal sampling to identify potential sources of pathogenic bacteria and to unravel at which stage of development dysbiosis- associated bacteria emerge. Results: Diseased individuals had drastically lower microbial alpha diversity and differed substantially in their microbial beta diversity from control individuals in all three regions of the gastrointestinal tract. -

Insights Into the Role of Erysipelotrichaceae in the Human Host
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Frontiers - Publisher Connector OPINION published: 20 November 2015 doi: 10.3389/fcimb.2015.00084 Insights into the Role of Erysipelotrichaceae in the Human Host Nadeem O. Kaakoush * School of Biotechnology and Biomolecular Sciences, Faculty of Science, University of New South Wales, Sydney, NSW, Australia Keywords: Erysipelotrichaceae, gastrointestinal, microbiota, metabolic disorders, inflammatory bowel diseases, colorectal cancer Understanding the human gut microbiota has garnered interest from researchers and clinicians to pharmaceutical companies looking at novel mechanisms to manipulate the microbiota for the benefit of the host. Studies on the gut microbiota can be loosely characterized into three areas that include investigating the microbiota’s role in the physiology of the healthy gut, in the establishment of gastrointestinal disease, and in extra-intestinal manifestations. With deep sequencing technologies now in routine use in the research environment, novel members of the gut microbiota are coming to light, and our understanding of this complex ecosystem and its relationship to the host is slowly improving. IMPORTANCE OF ERYSIPELOTRICHACEAE IN HUMANS Edited by: Reports documenting a potential role for the bacterial family Erysipelotrichaceae, which belongs D. Scott Merrell, to the Firmicutes phylum, in host physiology and/or disease are on the rise. However, more often Uniformed Services University of the than not, these organisms are mentioned in passing, despite the fact that members of this bacterial Health Sciences, USA family appear to be highly immunogenic and can potentially flourish post-treatment with broad Reviewed by: spectrum antibiotics (Zhao et al., 2013; Palm et al., 2014; Dinh et al., 2015). -

Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention
J Neurogastroenterol Motil, Vol. 26 No. 4 October, 2020 pISSN: 2093-0879 eISSN: 2093-0887 https://doi.org/10.5056/jnm20080 JNM Journal of Neurogastroenterology and Motility Original Article Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention Choong-Kyun Noh and Kwang Jae Lee* Department of Gastroenterology, Ajou University School of Medicine, Suwon, Gyeonggi-do, Korea Background/Aims The pathophysiology of functional abdominal bloating and distention (FABD) is unclear yet. Our aim is to compare the diversity and composition of fecal microbiota in patients with FABD and healthy individuals, and to evaluate the relationship between small intestinal bacterial overgrowth (SIBO) and dysbiosis. Methods The microbiota of fecal samples was analyzed from 33 subjects, including 12 healthy controls and 21 patients with FABD diagnosed by the Rome IV criteria. FABD patients underwent a hydrogen breath test. Fecal microbiota composition was determined by 16S ribosomal RNA amplification and sequencing. Results Overall fecal microbiota composition of the FABD group differed from that of the control group. Microbial diversity was significantly lower in the FABD group than in the control group. Significantly higher proportion of Proteobacteria and significantly lower proportion of Actinobacteria were observed in FABD patients, compared with healthy controls. Compared with healthy controls, significantly higher proportion of Faecalibacterium in FABD patients and significantly higher proportion of Prevotella and Faecalibacterium in SIBO (+) patients with FABD were found. Faecalibacterium prausnitzii, was significantly more abundant, but Bacteroides uniformis and Bifidobacterium adolescentis were significantly less abundant in patients with FABD, compared with healthy controls. Significantly more abundant Prevotella copri and F. -

New Insights in Ulcerative Colitis Associated Gut Microbiota in South American Population: Akkermansia and Collinsella, Two Dist
medRxiv preprint doi: https://doi.org/10.1101/2020.07.29.20164764; this version posted August 3, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . New insights in Ulcerative Colitis Associated Gut Microbiota in South American Population: Akkermansia and Collinsella, two distinctive genera found in Argentine subjects. 1,2 2,3 1,2,3 5 5 Rosso Ayelén * , Aguilera Pablo * , Quesada Sofía , Cerezo Jimena , Spiazzi Renata , 5 5 7,8,9 2,3 Conlon Carolina , Milano Claudia , Gregorio Iraola , Coluccio Leskow Federico , 1,2,3,6 1,2,3,4 Penas-Steinhardt Alberto , Belforte Fiorella S. . * Contributed equally 1 Laboratorio de Genómica Computacional (GeC-UNLu), Departamento de Ciencias Básicas, Universidad Nacional de Luján, Luján, Argentina. (http://gec.unlu.edu.ar) 2 Programa del Estudio de Comunicación y Señalización Interreino (PECSI-UNLu), Departamento de Ciencias Básicas, Universidad Nacional de Luján, Luján, Argentina. 3 Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), Argentina. 4 Instituto de Ecología y Desarrollo Sustentable (INEDES-CONICET-UNLu), Departamento de Ciencias Básicas, Universidad Nacional de Luján, Luján, Argentina. 5 Servicio de Gastroenterología, Hospital Nacional Prof. Alejandro Posadas, Buenos Aires, Argentina 6 Instituto Universitario de Ciencias de la Salud, Fundación H.A. Barceló, Ciudad Autónoma de Buenos Aires, Argentina 7 Laboratorio de Genómica Microbiana, Institut Pasteur Montevideo, Montevideo, Uruguay. 8 Centro de Biología Integrativa, Universidad Mayor, Santiago, Chile. 9 Wellcome Sanger Institute, Wellcome Genome Campus, Hinxton, United Kingdom. -

Bacteriophages As Potential New Mammalian Pathogens George V
www.nature.com/scientificreports OPEN Bacteriophages as potential new mammalian pathogens George V. Tetz 1, Kelly V. Ruggles2,3, Hua Zhou3, Adriana Heguy4,5,6, Aristotelis Tsirigos3,4,5 & Victor Tetz1 Received: 4 May 2017 Increased intestinal permeability and translocation of gut bacteria trigger various polyaetiological Accepted: 23 June 2017 diseases associated with chronic infammation and underlie a variety of poorly treatable pathologies. Published online: 1 August 2017 Previous studies have established a primary role of the microbiota composition and intestinal permeability in such pathologies. Using a rat model, we examined the efects of exposure to a bacteriophage cocktail on intestinal permeability and relative abundance of taxonomic units in the gut bacterial community. There was an increase in markers of impaired gut permeability, such as the lactulose/mannitol ratio, plasma endotoxin concentrations, and serum levels of infammation- related cytokines, following the bacteriophage challenge. We observed signifcant diferences in the alpha diversity of faecal bacterial species and found that richness and diversity index values increased following the bacteriophage challenge. There was a reduction in the abundance of Blautia, Catenibacterium, Lactobacillus, and Faecalibacterium species and an increase in Butyrivibrio, Oscillospira and Ruminococcus after bacteriophage administration. These fndings provide novel insights into the role of bacteriophages as potentially pathogenic for mammals and their possible implication in the development of diseases associated with increased intestinal permeability. Our knowledge of the intestinal microbiota has greatly expanded over the last decade, and growing evidence sug- gests that alterations of the intestinal microbiota are critical pathogenic factors that trigger various polyaetiologi- cal diseases associated with increased intestinal permeability and chronic infammation1–3. -
Emendation of Genus Collinsella and Proposal of Collinsella Stercoris Sp. Nov. and Collinsella Intestinalis Sp. Nov
International Journal of Systematic and Evolutionary Microbiology (2000), 50, 1767–1774 Printed in Great Britain Emendation of genus Collinsella and proposal of Collinsella stercoris sp. nov. and Collinsella intestinalis sp. nov. Akiko Kageyama and Yoshimi Benno Author for correspondence: Akiko Kageyama. Tel: j81 48 462 1111 ext. 5132. Fax: j81 48 462 4619. e-mail: kageyama!jcm.riken.go.jp Japan Collection of Collinsella aerofaciens-like strains isolated from human faeces were Microorganisms, The characterized by biochemical tests, cell wall murein analysis and 16S rDNA Institute of Physical and Chemical Research (RIKEN), analysis. The results indicated that these strains are phylogenetically a Wako, Saitama 351-0198, member of the family Coriobacteriaceae and close to the genus Collinsella. Japan Their phenotypic characters resembled those of Collinsella aerofaciens. Determination of DNA–DNA relatedness showed that these strains could be divided into two groups (groups 1 and 2). Collinsella aerofaciens and both new groups have A4-type cell wall murein. Based on their phenotypic and phylogenetic characters, two new species of the genus Collinsella are proposed for the isolated strains: Collinsella stercoris for group 1 and Collinsella intestinalis for group 2. Species-specific PCR primer sets for these two species were also constructed. Using these primer sets, Collinsella stercoris and Collinsella intestinalis can be identified easily and rapidly. Keywords: Collinsella stercoris sp. nov., Collinsella intestinalis sp. nov., 16S rDNA, cell wall, PCR INTRODUCTION transferred to other genera or new genera with specific phylogenetic characters (Kageyama et al., 1999a, b; Collinsella aerofaciens belongs to the family Corio- Ludwig et al., 1992; Wade et al., 1999). -

Improved Diagnostics and Further Investigation of Condemnations and Outbreaks Associated with Erysipelothrix Spp Joseph Samuel Bender Iowa State University
Iowa State University Capstones, Theses and Graduate Theses and Dissertations Dissertations 2010 Improved diagnostics and further investigation of condemnations and outbreaks associated with Erysipelothrix spp Joseph Samuel Bender Iowa State University Follow this and additional works at: https://lib.dr.iastate.edu/etd Part of the Veterinary Microbiology and Immunobiology Commons Recommended Citation Bender, Joseph Samuel, "Improved diagnostics and further investigation of condemnations and outbreaks associated with Erysipelothrix spp" (2010). Graduate Theses and Dissertations. 11370. https://lib.dr.iastate.edu/etd/11370 This Thesis is brought to you for free and open access by the Iowa State University Capstones, Theses and Dissertations at Iowa State University Digital Repository. It has been accepted for inclusion in Graduate Theses and Dissertations by an authorized administrator of Iowa State University Digital Repository. For more information, please contact [email protected]. Improved diagnostics and further investigation of condemnations and outbreaks associated with Erysipelothrix spp by Joseph Samuel Bender A thesis submitted to the graduate faculty in partial fulfillment of the requirements for the degree of MASTER OF SCIENCE Major: Veterinary Preventive Medicine Program of Study Committee: Tanja Opriessnig, Major Professor Kent J. Schwartz Leo L. Timms Iowa State University Ames, Iowa 2010 Copyright © Joseph Samuel Bender, 2010. All rights reserved. ii TABLE OF CONTENTS CHAPTER 1. GENERAL INTRODUCTION Thesis Organization 1 Statement of Problem and Research Summary 1 Reference List 3 CHAPTER 2. LITERATURE REVIEW Introduction 4 Morphology, Growth, and Biological Characteristics 5 Epidemiology 6 Prevalence and Condemnations 7 Pathogenesis and Clinical Signs of Disease 9 Virulence Factors 11 Diagnosis 14 Characterization 19 Prevention and Biologics Development 21 Reference List 23 CHAPTER 3. -

Erysipelothrix Rhusiopathiae in Laying Hens
Erysipelothrix rhusiopathiae in Laying Hens Helena Eriksson Faculty of Veterinary Medicine and Animal Science Department of Clinical Sciences Uppsala Doctoral Thesis Swedish University of Agricultural Sciences Uppsala 2013 Acta Universitatis agriculturae Sueciae 2013:26 Cover: “Ruvande”. Sculpture by Hermine Keller. (photo: H. Keller) ISSN 1652-6880 ISBN (Printed copy) 978-91-576-7790-7 ISBN (Electronic copy) 978-91-576-7791-4 © 2013 Helena Eriksson, Uppsala Print: SLU Service/Repro, Uppsala 2013 Erysipelothrix rhusiopathiae in Laying Hens Abstract The bacterium Erysipelothrix rhusiopathiae can infect a wide range of mammals (including humans) and birds. Disease outbreaks (erysipelas) have been considered unusual in chickens internationally, but outbreaks with high mortality and egg production losses have been diagnosed in Swedish laying hen flocks every year since 1998. Different aspects of E. rhusiopathiae infection in chickens were examined in this thesis with the aim of preventing future outbreaks. These aspects included determining occurrence of the bacterium in different housing systems for laying hens and the potential of the poultry red mite (Dermanyssus gallinae) to carry it, characterization of isolates of the bacterium from different hosts using pulsed-field gel electrophoresis (PFGE), serotyping, antimicrobial susceptibility testing and 16S rRNA gene sequencing, and determining the incidence of E. rhusiopathiae in the environment on affected organic laying hen farms by the use of selective culture and PCR. The results showed an association between erysipelas outbreaks and housing system. Flocks in free-range systems appeared to be at a higher risk than flocks in indoor litter- based systems, while flocks in cages appeared to be at the lowest risk.