Depression and Microbiome—Study on the Relation and Contiguity Between Dogs and Humans
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Uneven Distribution of Cobamide Biosynthesis and Dependence in Bacteria Predicted By
bioRxiv preprint doi: https://doi.org/10.1101/342006; this version posted June 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 1 Uneven distribution of cobamide biosynthesis and dependence in bacteria predicted by 2 comparative genomics 3 4 Running title: Cobamide biosynthesis predictions 5 6 Amanda N. Shelton1, Erica C. Seth1, Kenny C. Mok1, Andrew W. Han2, Samantha N. Jackson3,4, 7 David R. Haft3, Michiko E. Taga1* 8 9 1 Department of Plant & Microbial Biology, University of California, Berkeley, Berkeley, CA, 10 USA 11 2 Second Genome, Inc., South San Francisco, CA, USA 12 3 J. Craig Venter Institute, Rockville, MD, USA 13 4 Current address: Department of Biological & Environmental Engineering, Cornell University, 14 Ithaca, NY, USA 15 16 * Address correspondence to Michiko E. Taga, [email protected] 17 18 19 -- 20 Abstract 21 22 The vitamin B12 family of cofactors known as cobamides are essential for a variety of microbial 23 metabolisms. We used comparative genomics of 11,000 bacterial species to analyze the extent 1 bioRxiv preprint doi: https://doi.org/10.1101/342006; this version posted June 8, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC 4.0 International license. 24 and distribution of cobamide production and use across bacteria. -

Supplementary Information
doi: 10.1038/nature06269 SUPPLEMENTARY INFORMATION METAGENOMIC AND FUNCTIONAL ANALYSIS OF HINDGUT MICROBIOTA OF A WOOD FEEDING HIGHER TERMITE TABLE OF CONTENTS MATERIALS AND METHODS 2 • Glycoside hydrolase catalytic domains and carbohydrate binding modules used in searches that are not represented by Pfam HMMs 5 SUPPLEMENTARY TABLES • Table S1. Non-parametric diversity estimators 8 • Table S2. Estimates of gross community structure based on sequence composition binning, and conserved single copy gene phylogenies 8 • Table S3. Summary of numbers glycosyl hydrolases (GHs) and carbon-binding modules (CBMs) discovered in the P3 luminal microbiota 9 • Table S4. Summary of glycosyl hydrolases, their binning information, and activity screening results 13 • Table S5. Comparison of abundance of glycosyl hydrolases in different single organism genomes and metagenome datasets 17 • Table S6. Comparison of abundance of glycosyl hydrolases in different single organism genomes (continued) 20 • Table S7. Phylogenetic characterization of the termite gut metagenome sequence dataset, based on compositional phylogenetic analysis 23 • Table S8. Counts of genes classified to COGs corresponding to different hydrogenase families 24 • Table S9. Fe-only hydrogenases (COG4624, large subunit, C-terminal domain) identified in the P3 luminal microbiota. 25 • Table S10. Gene clusters overrepresented in termite P3 luminal microbiota versus soil, ocean and human gut metagenome datasets. 29 • Table S11. Operational taxonomic unit (OTU) representatives of 16S rRNA sequences obtained from the P3 luminal fluid of Nasutitermes spp. 30 SUPPLEMENTARY FIGURES • Fig. S1. Phylogenetic identification of termite host species 38 • Fig. S2. Accumulation curves of 16S rRNA genes obtained from the P3 luminal microbiota 39 • Fig. S3. Phylogenetic diversity of P3 luminal microbiota within the phylum Spirocheates 40 • Fig. -

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

Supporting Information
Supporting Information Lozupone et al. 10.1073/pnas.0807339105 SI Methods nococcus, and Eubacterium grouped with members of other Determining the Environmental Distribution of Sequenced Genomes. named genera with high bootstrap support (Fig. 1A). One To obtain information on the lifestyle of the isolate and its reported member of the Bacteroidetes (Bacteroides capillosus) source, we looked at descriptive information from NCBI grouped firmly within the Firmicutes. This taxonomic error was (www.ncbi.nlm.nih.gov/genomes/lproks.cgi) and other related not surprising because gut isolates have often been classified as publications. We also determined which 16S rRNA-based envi- Bacteroides based on an obligate anaerobe, Gram-negative, ronmental surveys of microbial assemblages deposited near- nonsporulating phenotype alone (6, 7). A more recent 16S identical sequences in GenBank. We first downloaded the gbenv rRNA-based analysis of the genus Clostridium defined phylo- files from the NCBI ftp site on December 31, 2007, and used genetically related clusters (4, 5), and these designations were them to create a BLAST database. These files contain GenBank supported in our phylogenetic analysis of the Clostridium species in the HGMI pipeline. We thus designated these Clostridium records for the ENV database, a component of the nonredun- species, along with the species from other named genera that dant nucleotide database (nt) where 16S rRNA environmental cluster with them in bootstrap supported nodes, as being within survey data are deposited. GenBank records for hits with Ͼ98% these clusters. sequence identity over 400 bp to the 16S rRNA sequence of each of the 67 genomes were parsed to get a list of study titles Annotation of GTs and GHs. -

Product Sheet Info
Product Information Sheet for HM-289 Facklamia sp., Strain HGF4 Incubation: Temperature: 37°C Catalog No. HM-289 Atmosphere: Aerobic Propagation: 1. Keep vial frozen until ready for use, then thaw. For research use only. Not for human use. 2. Transfer the entire thawed aliquot into a single tube of broth. Contributor: 3. Use several drops of the suspension to inoculate an Thomas M. Schmidt, Professor, Department of Microbiology agar slant and/or plate. and Molecular Genetics, Michigan State University, East 4. Incubate the tube, slant and/or plate at 37°C for 48 to Lansing, Michigan, USA 168 hours. Manufacturer: Citation: BEI Resources Acknowledgment for publications should read “The following reagent was obtained through BEI Resources, NIAID, NIH as Product Description: part of the Human Microbiome Project: Facklamia sp., Strain Bacteria Classification: Aerococcaceae, Facklamia HGF4, HM-289.” Species: Facklamia sp. Strain: HGF4 Biosafety Level: 2 Original Source: Facklamia sp., strain HGF4 is a human Appropriate safety procedures should always be used with gastrointestinal isolate.1 this material. Laboratory safety is discussed in the following Comments: Facklamia sp., strain HGF4 (HMP ID 9411) is a publication: U.S. Department of Health and Human Services, reference genome for The Human Microbiome Project Public Health Service, Centers for Disease Control and (HMP). HMP is an initiative to identify and characterize Prevention, and National Institutes of Health. Biosafety in human microbial flora. The complete genome of Facklamia Microbiological and Biomedical Laboratories. 5th ed. sp., strain HGF4 is currently being sequenced at the J. Washington, DC: U.S. Government Printing Office, 2009; see Craig Venter Institute. -

Aerococcus Viridans: a Rare Pathogen Causing Urinary Tract Infection Microbiology Section Microbiology
DOI: 10.7860/JCDR/2017/23997.9229 Case Series Aerococcus Viridans: A Rare Pathogen Causing Urinary Tract Infection Microbiology Section Microbiology BALVINDER MOHAN1, KAMRAN ZAMAN2, NAVEEN ANAND3, NEELAM TANEJA4 ABSTRACT Aerococci are Gram-positive cocci with colony morphology similar to viridans streptococci. Most often these isolates in clinical samples are misidentified and considered insignificant. However, with the use newer techniques like Matrix-Assisted Laser Desorption Ionization Time-of-Flight Mass-Spectrometry (MALDI-TOF MS), aerococci have been recognized as significant human pathogens capable of causing a diverse spectrum of infections. Among the different species of aerococci, Aerococcus urinae is the most common agent causing Urinary Tract Infection (UTI) followed by A. sanguinocola. Aerococcus viridans (A. viridans) have been reported rarely in urinary tract infections. The antimicrobial resistance in aerococci in terms of its intrinsic resistance and evolving resistance to penicillin and vancomycin has raised the concern for better understanding of this pathogen. We recently encountered two cases of nosocomial UTI caused by A. viridans which are being reported here. Keywords: Aerococci, Nosocomial, Vancomycin UTI form a major component of the most commonly encountered The present case reports have been retrospectively reviewed and bacterial infections in routine clinical practice. Most of these reported and it did not require any institutional ethics committee infections are caused by members of the Enterobacteriaceae family; approval. The patients involved in this report have given their written in particular, Escherichia coli and the Gram-positive cocci such as informed consent authorizing use and disclosure of their protected Staphylococcus spp and Enterococcus spp [1]. In routine clinical health information. -

WO 2018/064165 A2 (.Pdf)
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/064165 A2 05 April 2018 (05.04.2018) W !P O PCT (51) International Patent Classification: Published: A61K 35/74 (20 15.0 1) C12N 1/21 (2006 .01) — without international search report and to be republished (21) International Application Number: upon receipt of that report (Rule 48.2(g)) PCT/US2017/053717 — with sequence listing part of description (Rule 5.2(a)) (22) International Filing Date: 27 September 2017 (27.09.2017) (25) Filing Language: English (26) Publication Langi English (30) Priority Data: 62/400,372 27 September 2016 (27.09.2016) US 62/508,885 19 May 2017 (19.05.2017) US 62/557,566 12 September 2017 (12.09.2017) US (71) Applicant: BOARD OF REGENTS, THE UNIVERSI¬ TY OF TEXAS SYSTEM [US/US]; 210 West 7th St., Austin, TX 78701 (US). (72) Inventors: WARGO, Jennifer; 1814 Bissonnet St., Hous ton, TX 77005 (US). GOPALAKRISHNAN, Vanch- eswaran; 7900 Cambridge, Apt. 10-lb, Houston, TX 77054 (US). (74) Agent: BYRD, Marshall, P.; Parker Highlander PLLC, 1120 S. Capital Of Texas Highway, Bldg. One, Suite 200, Austin, TX 78746 (US). (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -

Development of Bacterial Communities in Biological Soil Crusts Along
1 Development of bacterial communities in biological soil crusts along 2 a revegetation chronosequence in the Tengger Desert, northwest 3 China 4 5 Author names and affiliations: 6 Lichao Liu1, Yubing Liu1, 2 *, Peng Zhang1, Guang Song1, Rong Hui1, Zengru Wang1, Jin Wang1, 2 7 1Shapotou Desert Research & Experiment Station, Northwest Institute of Eco-Environment and Resources, Chinese 8 Academy of Sciences, Lanzhou, 730000, China 9 2Key Laboratory of Stress Physiology and Ecology in Cold and Arid Regions of Gansu Province, Northwest Institute 10 of Eco–Environment and Resources, Chinese Academy of Sciences, Lanzhou 730000, China 11 12 * Corresponding author: Yubing Liu 13 Address: Donggang West Road 320, Lanzhou 730000, P. R. China. 14 Tel: +86 0931 4967202. 15 E-mail address: [email protected] 16 17 Abstract. Knowledge of structure and function of microbial communities in different 18 successional stages of biological soil crusts (BSCs) is still scarce for desert areas. In this study, 19 Illumina MiSeq sequencing was used to assess the composition changes of bacterial communities 20 in different ages of BSCs in the revegetation of Shapotou in the Tengger Desert. The most dominant 21 phyla of bacterial communities shifted with the changed types of BSCs in the successional stages, 22 from Firmicutes in mobile sand and physical crusts to Actinobacteria and Proteobacteria in BSCs, 23 and the most dominant genera shifted from Bacillus, Enterococcus and Lactococcus to 24 RB41_norank and JG34-KF-361_norank. Alpha diversity and quantitative real-time PCR analysis 25 indicated that bacteria richness and abundance reached their highest levels after 15 years of BSC 26 development. -

A Metagenomic Β-Glucuronidase Uncovers a Core Adaptive Function of the Human Intestinal Microbiome
A metagenomic β-glucuronidase uncovers a core adaptive function of the human intestinal microbiome Karine Gloux, Olivier Berteau, Hanane El oumami, Fabienne Béguet, Marion Leclerc, and Joël Doré1 Institut National de la Recherche Agronomique, Unité Mixte de Recherche 1319 Micalis, F-78352 Jouy en Josas, France Edited by Todd R. Klaenhammer, North Carolina State University, Raleigh, NC, and approved June 1, 2010 (received for review February 4, 2010) In the human gastrointestinal tract, bacterial β-D-glucuronidases (BG; XIVa), major reservoirs of BG-positive bacteria (11), are dys- E.C. 3.2.1.31) are involved both in xenobiotic metabolism and in some biosis markers in Crohn’s disease (12–15) and in intestinal carci- of the beneficial effects of dietary compounds. Despite their biolog- nogenesis (16). Nevertheless, the relative contribution of bacterial ical significance, investigations are hampered by the fact that only strains to the global intestinal BG activity remains unclear, and a few BGs have so far been studied. A functional metagenomic the molecular bases are essentially unknown. approach was therefore performed on intestinal metagenomic In the human digestive tract, bacterial BGs are known to be libraries using chromogenic glucuronides as probes. Using this strat- distributed among the Enterobacteriaceae family in some Firmi- egy, 19 positive metagenomic clones were identified but only one cutes genera (Lactobacillus, Streptococcus, Clostridium, Rumino- exhibited strong β-D-glucuronidase activity when subcloned into an coccus, Roseburia, and Faecalibacterium) and in a specific species expression vector. The cloned gene encoded a β-D-glucuronidase of Actinobacteria (Bifidobacterium dentium) (11, 17, 18). On (called H11G11-BG) that had distant amino acid sequence homolo- the basis of their sequence, BGs are highly homologous to gies and an additional C terminus domain compared with known β-galactosidases and only a few of them have been investigated β fi -D-glucuronidases. -
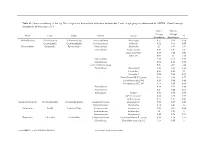
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Multi-Product Lactic Acid Bacteria Fermentations: a Review
fermentation Review Multi-Product Lactic Acid Bacteria Fermentations: A Review José Aníbal Mora-Villalobos 1 ,Jéssica Montero-Zamora 1, Natalia Barboza 2,3, Carolina Rojas-Garbanzo 3, Jessie Usaga 3, Mauricio Redondo-Solano 4, Linda Schroedter 5, Agata Olszewska-Widdrat 5 and José Pablo López-Gómez 5,* 1 National Center for Biotechnological Innovations of Costa Rica (CENIBiot), National Center of High Technology (CeNAT), San Jose 1174-1200, Costa Rica; [email protected] (J.A.M.-V.); [email protected] (J.M.-Z.) 2 Food Technology Department, University of Costa Rica (UCR), San Jose 11501-2060, Costa Rica; [email protected] 3 National Center for Food Science and Technology (CITA), University of Costa Rica (UCR), San Jose 11501-2060, Costa Rica; [email protected] (C.R.-G.); [email protected] (J.U.) 4 Research Center in Tropical Diseases (CIET) and Food Microbiology Section, Microbiology Faculty, University of Costa Rica (UCR), San Jose 11501-2060, Costa Rica; [email protected] 5 Bioengineering Department, Leibniz Institute for Agricultural Engineering and Bioeconomy (ATB), 14469 Potsdam, Germany; [email protected] (L.S.); [email protected] (A.O.-W.) * Correspondence: [email protected]; Tel.: +49-(0331)-5699-857 Received: 15 December 2019; Accepted: 4 February 2020; Published: 10 February 2020 Abstract: Industrial biotechnology is a continuously expanding field focused on the application of microorganisms to produce chemicals using renewable sources as substrates. Currently, an increasing interest in new versatile processes, able to utilize a variety of substrates to obtain diverse products, can be observed. -

Berberine Alters Gut Microbial Function Through Modulation of Bile Acids Patricia G
Wolf et al. BMC Microbiology (2021) 21:24 https://doi.org/10.1186/s12866-020-02020-1 RESEARCH ARTICLE Open Access Berberine alters gut microbial function through modulation of bile acids Patricia G. Wolf1,2,3,4,5, Saravanan Devendran3,5,6, Heidi L. Doden3,5, Lindsey K. Ly3,4,5, Tyler Moore7, Hajime Takei8, Hiroshi Nittono8, Tsuyoshi Murai9, Takao Kurosawa9, George E. Chlipala10, Stefan J. Green10, Genta Kakiyama11, Purna Kashyap12, Vance J. McCracken13, H. Rex Gaskins3,4,5,14,15, Patrick M. Gillevet6 and Jason M. Ridlon3,4,5,15,16* Abstract Background: Berberine (BBR) is a plant-based nutraceutical that has been used for millennia to treat diarrheal infections and in contemporary medicine to improve patient lipid profiles. Reduction in lipids, particularly cholesterol, is achieved partly through up-regulation of bile acid synthesis and excretion into the gastrointestinal tract (GI). The efficacy of BBR is also thought to be dependent on structural and functional alterations of the gut microbiome. However, knowledge of the effects of BBR on gut microbiome communities is currently lacking. Distinguishing indirect effects of BBR on bacteria through altered bile acid profiles is particularly important in understanding how dietary nutraceuticals alter the microbiome. Results: Germfree mice were colonized with a defined minimal gut bacterial consortium capable of functional bile acid metabolism (Bacteroides vulgatus, Bacteroides uniformis, Parabacteroides distasonis, Bilophila wadsworthia, Clostridium hylemonae, Clostridium hiranonis, Blautia producta; B4PC2). Multi-omics (bile acid metabolomics, 16S rDNA sequencing, cecal metatranscriptomics) were performed in order to provide a simple in vivo model from which to identify network-based correlations between bile acids and bacterial transcripts in the presence and absence of dietary BBR.