Impact of Diet and Individual Variation on Intestinal Microbiota Composition and Fermentation Products in Obese Men
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio In
microorganisms Review The Influence of Probiotics on the Firmicutes/Bacteroidetes Ratio in the Treatment of Obesity and Inflammatory Bowel disease Spase Stojanov 1,2, Aleš Berlec 1,2 and Borut Štrukelj 1,2,* 1 Faculty of Pharmacy, University of Ljubljana, SI-1000 Ljubljana, Slovenia; [email protected] (S.S.); [email protected] (A.B.) 2 Department of Biotechnology, Jožef Stefan Institute, SI-1000 Ljubljana, Slovenia * Correspondence: borut.strukelj@ffa.uni-lj.si Received: 16 September 2020; Accepted: 31 October 2020; Published: 1 November 2020 Abstract: The two most important bacterial phyla in the gastrointestinal tract, Firmicutes and Bacteroidetes, have gained much attention in recent years. The Firmicutes/Bacteroidetes (F/B) ratio is widely accepted to have an important influence in maintaining normal intestinal homeostasis. Increased or decreased F/B ratio is regarded as dysbiosis, whereby the former is usually observed with obesity, and the latter with inflammatory bowel disease (IBD). Probiotics as live microorganisms can confer health benefits to the host when administered in adequate amounts. There is considerable evidence of their nutritional and immunosuppressive properties including reports that elucidate the association of probiotics with the F/B ratio, obesity, and IBD. Orally administered probiotics can contribute to the restoration of dysbiotic microbiota and to the prevention of obesity or IBD. However, as the effects of different probiotics on the F/B ratio differ, selecting the appropriate species or mixture is crucial. The most commonly tested probiotics for modifying the F/B ratio and treating obesity and IBD are from the genus Lactobacillus. In this paper, we review the effects of probiotics on the F/B ratio that lead to weight loss or immunosuppression. -

Effects of Dietary Supplementation with Hainanmycin on Protein Degradation and Populations of Ammonia-Producing Bacteria in Vitro
668 Asian Australas. J. Anim. Sci. Vol. 26, No. 5 : 668-674 May 2013 http://dx.doi.org/10.5713/ajas.2012.12589 www.ajas.info pISSN 1011-2367 eISSN 1976-5517 Effects of Dietary Supplementation with Hainanmycin on Protein Degradation and Populations of Ammonia-producing Bacteria In vitro Z. B. Wang1,2,a, H. S. Xin1,a, M. J. Wang1, Z. Y. Li1, Y. L. Qu2, S. J. Miao2 and Y. G. Zhang1,* 1 College of Animal Science and Technology, Northeast Agricultural University, Harbin, 150030, Heilongjiang, China ABSTRACT: An in vitro fermentation was conducted to determine the effects of hainanmycin on protein degradation and populations of ammonia-producing bacteria. The substrates (DM basis) for in vitro fermentation consisted of alfalfa hay (31.7%), Chinese wild rye grass hay (28.3%), ground corn grain (24.5%), soybean meal (15.5%) with a forage: concentrate of 60:40. Treatments were the control (no additive) and hainanmycin supplemented at 0.1 (H0.1), 1 (H1), 10 (H10), and 100 mg/kg (H100) of the substrates. After 24 h of fermentation, the highest addition level of hainanmycin decreased total VFA concentration and increased the final pH. The high addition level of hainanmycin (H1, H10, and H100) reduced (p<0.05) branched-chain VFA concentration, the molar proportion of acetate and butyrate, and ratio of acetate to propionate; and increased the molar proportion of propionate, except that for H1 the in molar proportion of acetate and isobutyrate was not changed (p>0.05). After 24 h of fermentation, H10 and H100 increased (p<0.05) concentrations of peptide nitrogen and AA nitrogen and proteinase activity, and decreased (p<0.05) NH3-N concentration and deaminase activity compared with control. -

Comparison of Enzymatic Activities and Proteomic Profiles Of
Sechovcová et al. Proteome Science (2019) 17:2 https://doi.org/10.1186/s12953-019-0150-3 RESEARCH Open Access Comparison of enzymatic activities and proteomic profiles of Butyrivibrio fibrisolvens grown on different carbon sources Hana Sechovcová1,5* , Lucie Kulhavá2,4, Kateřina Fliegerová1, Mária Trundová3, Daniel Morais6, Jakub Mrázek1 and Jan Kopečný1 Abstract Background: The rumen microbiota is one of the most complex consortia of anaerobes, involving archaea, bacteria, protozoa, fungi and phages. They are very effective at utilizing plant polysaccharides, especially cellulose and hemicelluloses. The most important hemicellulose decomposers are clustered with the genus Butyrivibrio.As the related species differ in their range of hydrolytic activities and substrate preferences, Butyrivibrio fibrisolvens was selected as one of the most effective isolates and thus suitable for proteomic studies on substrate comparisons in the extracellular fraction. The B. fibrisolvens genome is the biggest in the butyrivibria cluster and is focused on “environmental information processing” and “carbohydrate metabolism”. Methods: The study of the effect of carbon source on B. fibrisolvens 3071 was based on cultures grown on four substrates: xylose, glucose, xylan, xylan with 25% glucose. The enzymatic activities were studied by spectrophotometric and zymogram methods. Proteomic study was based on genomics, 2D electrophoresis and nLC/MS (Bruker Daltonics) analysis. Results: Extracellular β-endoxylanase as well as xylan β-xylosidase activities were induced with xylan. The presence of the xylan polymer induced hemicellulolytic enzymes and increased the protein fraction in the interval from 40 to 80 kDa. 2D electrophoresis with nLC/MS analysis of extracellular B. fibrisolvens 3071 proteins found 14 diverse proteins with significantly different expression on the tested substrates. -

Evaluation of 16S Rrna Primer Sets for Characterisation of Microbiota in Paediatric Patients with Autism Spectrum Disorder L
www.nature.com/scientificreports OPEN Evaluation of 16S rRNA primer sets for characterisation of microbiota in paediatric patients with autism spectrum disorder L. Palkova1,2, A. Tomova3, G. Repiska3, K. Babinska3, B. Bokor4, I. Mikula5, G. Minarik2, D. Ostatnikova3 & K. Soltys4,6* Abstract intestinal microbiota is becoming a signifcant marker that refects diferences between health and disease status also in terms of gut-brain axis communication. Studies show that children with autism spectrum disorder (ASD) often have a mix of gut microbes that is distinct from the neurotypical children. Various assays are being used for microbiota investigation and were considered to be universal. However, newer studies showed that protocol for preparing DNA sequencing libraries is a key factor infuencing results of microbiota investigation. The choice of DNA amplifcation primers seems to be the crucial for the outcome of analysis. In our study, we have tested 3 primer sets to investigate diferences in outcome of sequencing analysis of microbiota in children with ASD. We found out that primers detected diferent portion of bacteria in samples especially at phylum level; signifcantly higher abundance of Bacteroides and lower Firmicutes were detected using 515f/806r compared to 27f/1492r and 27f*/1495f primers. So, the question is whether a gold standard of Firmicutes/Bacteroidetes ratio is a valuable and reliable universal marker, since two primer sets towards 16S rRNA can provide opposite information. Moreover, signifcantly higher relative abundance of Proteobacteria was detected using 27f/1492r. The beta diversity of sample groups difered remarkably and so the number of observed bacterial genera. An advent of massively parallel sequencing (MPS) technologies has revolutionized an investigating of human microbiota1. -

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

Biotechnological Approaches to Manure Nutrient Management
COUNCIL FORFOR AGRICULTURALAGRICULTURAL SCIENCE SCIENCE AND AND TECHNOLOGY—1 TECHNOLOGY NUMBER 33 JULY 2006 ANIMAL AGRICULTURE'S FUTURE THROUGH BIOTECHNOLOGY, PART 4 BIOTECHNOLOGICAL APPROACHES TO MANURE NUTRIENT MANAGEMENT TASK FORCE MEMBERS: Xingen Lei, Chair, Department of Animal Science, Cornell Univer- neering. This type of technol- INTRODUCTION sity, Ithaca, New York; John P. Blake, Depart- Food animals are fed and ogy may be applied to de- produced for the purpose of ment of Poultry Science, Auburn University, crease total manure mass or feeding humans. Manure from Auburn, Alabama; Cecil W. Forsberg, Depart- concentrations of P, N, am- these animals is a valuable ment of Microbiology, University of Guelph, monia (NH3), trace elements, source of fertilizer, but con- Ontario, Canada; Danny G. Fox, Department of and other factors. Targeted modifications centrations of manure nutri- Animal Science, Cornell University, Ithaca, New 1 can be based strategically on ents such as phosphorus (P) , York; Elizabeth Grabau, Department of Plant nitrogen (N), and metals may plants, animals, ruminal and Pathology, Physiology, and Weed Science, Vir- exceed needs for plant growth intestinal microorganisms, and cause environmental pol- ginia Tech, Blacksburg; Zdzislaw Mroz, Ani- and diets. The plant-based lution. Thus, managing live- mal Sciences Group, Division of Nutrition and approaches include genetic stock manure nutrients has Food, Wageningen, The Netherlands; Alan L. and chemical modifications of been a task shared by those Sutton, Department of Animal Sciences, Purdue feeds (e.g., overexpressing hydrolytic enzymes such as interested in animal nutrition, University, West Lafayette, Indiana; William R. agronomy, and environmental phytase in seeds, decreasing Walker, DPI Global, Fort Dodge, Iowa; Ken- protection. -

WO 2018/064165 A2 (.Pdf)
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2018/064165 A2 05 April 2018 (05.04.2018) W !P O PCT (51) International Patent Classification: Published: A61K 35/74 (20 15.0 1) C12N 1/21 (2006 .01) — without international search report and to be republished (21) International Application Number: upon receipt of that report (Rule 48.2(g)) PCT/US2017/053717 — with sequence listing part of description (Rule 5.2(a)) (22) International Filing Date: 27 September 2017 (27.09.2017) (25) Filing Language: English (26) Publication Langi English (30) Priority Data: 62/400,372 27 September 2016 (27.09.2016) US 62/508,885 19 May 2017 (19.05.2017) US 62/557,566 12 September 2017 (12.09.2017) US (71) Applicant: BOARD OF REGENTS, THE UNIVERSI¬ TY OF TEXAS SYSTEM [US/US]; 210 West 7th St., Austin, TX 78701 (US). (72) Inventors: WARGO, Jennifer; 1814 Bissonnet St., Hous ton, TX 77005 (US). GOPALAKRISHNAN, Vanch- eswaran; 7900 Cambridge, Apt. 10-lb, Houston, TX 77054 (US). (74) Agent: BYRD, Marshall, P.; Parker Highlander PLLC, 1120 S. Capital Of Texas Highway, Bldg. One, Suite 200, Austin, TX 78746 (US). (81) Designated States (unless otherwise indicated, for every kind of national protection available): AE, AG, AL, AM, AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DJ, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IR, IS, JO, JP, KE, KG, KH, KN, KP, KR, KW, KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. -
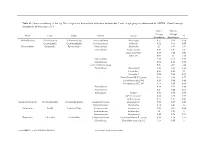
Genus Contributing to the Top 70% of Significant Dissimilarity of Bacteria Between Day 7 and 18 Age Groups As Determined by SIMPER
Table S1: Genus contributing to the top 70% of significant dissimilarity of bacteria between day 7 and 18 age groups as determined by SIMPER. Overall average dissimilarity between ages is 51%. Day 7 Day 18 Average Average Phyla Class Order Family Genus % Abundance Abundance Actinobacteria Actinobacteria Actinomycetales Actinomycetaceae Actinomyces 0.57 0.24 0.74 Coriobacteriia Coriobacteriales Coriobacteriaceae Collinsella 0.51 0.61 0.57 Bacteroidetes Bacteroidia Bacteroidales Bacteroidaceae Bacteroides 2.1 1.63 1.03 Marinifilaceae Butyricimonas 0.82 0.81 0.9 Sanguibacteroides 0.08 0.42 0.59 CAG-873 0.01 1.1 1.68 Marinifilaceae 0.38 0.78 0.91 Marinifilaceae 0.78 1.04 0.97 p-2534-18B5 gut group 0.06 0.7 1.04 Prevotellaceae Alloprevotella 0.46 0.83 0.89 Prevotella 2 0.95 1.11 1.3 Prevotella 7 0.22 0.42 0.56 Prevotellaceae NK3B31 group 0.62 0.68 0.77 Prevotellaceae UCG-003 0.26 0.64 0.89 Prevotellaceae UCG-004 0.11 0.48 0.68 Prevotellaceae 0.64 0.52 0.89 Prevotellaceae 0.27 0.42 0.55 Rikenellaceae Alistipes 0.39 0.62 0.77 dgA-11 gut group 0.04 0.38 0.56 RC9 gut group 0.79 1.17 0.95 Epsilonbacteraeota Campylobacteria Campylobacterales Campylobacteraceae Campylobacter 0.58 0.83 0.97 Helicobacteraceae Helicobacter 0.12 0.41 0.6 Firmicutes Bacilli Lactobacillales Enterococcaceae Enterococcus 0.36 0.31 0.64 Lactobacillaceae Lactobacillus 1.4 1.24 1 Streptococcaceae Streptococcus 0.82 0.58 0.53 Firmicutes Clostridia Clostridiales Christensenellaceae Christensenellaceae R-7 group 0.38 1.36 1.56 Clostridiaceae Clostridium sensu stricto 1 1.16 0.58 -

Targeting the Gut Microbiome in Allogeneic Hematopoietic Stem Cell Transplantation
medRxiv preprint doi: https://doi.org/10.1101/2020.04.08.20058198; this version posted June 9, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted medRxiv a license to display the preprint in perpetuity. It is made available under a CC-BY-NC-ND 4.0 International license . Targeting the gut microbiome in allogeneic hematopoietic stem cell transplantation Marcel A. de Leeuw & Manuel X. Duval, GeneCreek List of Figures Contents 1 GM composition evolution across allo-HSCT . 2 I 2 Baseline GM composition and conditioning level . 3 NTRODUCTION 1 3 Top 10 variable importances estimated by the ran- dom survival forest models .............. 3 MATERIALS & METHODS 2 4 Biological safety level and aGvHD at onset . 3 DATA ANALYSIS .................. 2 5 Relative importance of regressors explaining the RESULTS 2 aGvHD status ...................... 3 OVERALL GM COMPOSITION EVOLUTION ACROSS 6 Co-exclusion by and co-occurrence with QPS species 4 ALLO-HSCT ................. 2 List of Tables CORRELATION BETWEEN CONDITIONING AND THE GM 2 BASELINE GM COMPOSITION AND SURVIVAL . 3 1 Prospective data sets used in the study . 1 AGVHD CASES, CONTROLS AND GM COMPOSITION 3 IMMUNO-MODULATING METABOLITES . 4 IN SILICO SCREENING OF THE ALLO-HSCT GM . 4 DISCUSSION 4 CONCLUSIONS 6 SUMMARY 6 DECLARATIONS 6 BIBLIOGRAPHY 7 NOTE: This preprint reports new research that has not been certified by peer review and should not be used to guide clinical practice. Revised manuscript medRxiv preprint doi: https://doi.org/10.1101/2020.04.08.20058198; this version posted June 9, 2020. -

Metagenomic Surveys of Gut Microbiota
Accepted Manuscript Metagenomic Surveys of Gut Microbiota Rahul Shubhra Mandal, Sudipto Saha, Santasabuj Das PII: S1672-0229(15)00054-6 DOI: http://dx.doi.org/10.1016/j.gpb.2015.02.005 Reference: GPB 159 To appear in: Genomics, Proteomics & Bioinformatics Received Date: 8 July 2014 Revised Date: 10 February 2015 Accepted Date: 26 February 2015 Please cite this article as: R.S. Mandal, S. Saha, S. Das, Metagenomic Surveys of Gut Microbiota, Genomics, Proteomics & Bioinformatics (2015), doi: http://dx.doi.org/10.1016/j.gpb.2015.02.005 This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain. 1 Metagenomic Surveys of Gut Microbiota 2 3 Rahul Shubhra Mandal1,a, Sudipto Saha2,*,b, Santasabuj Das1,3,*,c 4 5 1Biomedical Informatics Centre, National Institute of Cholera and Enteric Diseases, Kolkata 6 700010, India 7 2Bioinformatics Centre, Bose Institute, Kolkata 700054, India 8 3Division of Clinical Medicine, National Institute of Cholera and Enteric Diseases, Kolkata 9 700010, India 10 11 *Corresponding authors. 12 E-mail: [email protected] (Das S), [email protected] (Saha S). 13 Running title: Mandal RS et al / Metagenomic Analysis of Gut Microbiota 14 15 aORCID: 0000-0001-8939-3362. -

Dietary Supplementation with Inulin-Propionate Ester Or Inulin
Gut microbiota ORIGINAL ARTICLE Dietary supplementation with inulin-propionate ester Gut: first published as 10.1136/gutjnl-2019-318424 on 10 April 2019. Downloaded from or inulin improves insulin sensitivity in adults with overweight and obesity with distinct effects on the gut microbiota, plasma metabolome and systemic inflammatory responses: a randomised cross- over trial Edward S Chambers, 1 Claire S Byrne,1 Douglas J Morrison,2 Kevin G Murphy,3 Tom Preston,2 Catriona Tedford,4 Isabel Garcia-Perez,5 Sofia Fountana,5 Jose Ivan Serrano-Contreras,5 Elaine Holmes,5 Catherine J Reynolds,6 Jordie F Roberts,6 Rosemary J Boyton,6 Daniel M Altmann,6 Julie A K McDonald, 7 Julian R Marchesi,7,8 Arne N Akbar,9 Natalie E Riddell,10 Gareth A Wallis,11 Gary S Frost1 ► Additional material is ABSTRact published online only. To view Objective To investigate the underlying mechanisms Significance of this study please visit the journal online behind changes in glucose homeostasis with delivery (http:// dx. doi. org/ 10. 1136/ What is already known on this subject? gutjnl- 2019- 318424). of propionate to the human colon by comprehensive and coordinated analysis of gut bacterial composition, ► Short-chain fatty acids (SCFA), derived from For numbered affiliations see fermentation of dietary fibre by the gut end of article. plasma metabolome and immune responses. Design Twelve non-diabetic adults with overweight microbiota, have been shown to improve host insulin sensitivity. Correspondence to and obesity received 20 g/day of inulin-propionate ester (IPE), -

Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention
J Neurogastroenterol Motil, Vol. 26 No. 4 October, 2020 pISSN: 2093-0879 eISSN: 2093-0887 https://doi.org/10.5056/jnm20080 JNM Journal of Neurogastroenterology and Motility Original Article Fecal Microbiota Alterations and Small Intestinal Bacterial Overgrowth in Functional Abdominal Bloating/Distention Choong-Kyun Noh and Kwang Jae Lee* Department of Gastroenterology, Ajou University School of Medicine, Suwon, Gyeonggi-do, Korea Background/Aims The pathophysiology of functional abdominal bloating and distention (FABD) is unclear yet. Our aim is to compare the diversity and composition of fecal microbiota in patients with FABD and healthy individuals, and to evaluate the relationship between small intestinal bacterial overgrowth (SIBO) and dysbiosis. Methods The microbiota of fecal samples was analyzed from 33 subjects, including 12 healthy controls and 21 patients with FABD diagnosed by the Rome IV criteria. FABD patients underwent a hydrogen breath test. Fecal microbiota composition was determined by 16S ribosomal RNA amplification and sequencing. Results Overall fecal microbiota composition of the FABD group differed from that of the control group. Microbial diversity was significantly lower in the FABD group than in the control group. Significantly higher proportion of Proteobacteria and significantly lower proportion of Actinobacteria were observed in FABD patients, compared with healthy controls. Compared with healthy controls, significantly higher proportion of Faecalibacterium in FABD patients and significantly higher proportion of Prevotella and Faecalibacterium in SIBO (+) patients with FABD were found. Faecalibacterium prausnitzii, was significantly more abundant, but Bacteroides uniformis and Bifidobacterium adolescentis were significantly less abundant in patients with FABD, compared with healthy controls. Significantly more abundant Prevotella copri and F.