ZODIAC: Database-Independent Molecular Formula Annotation Using Gibbs Sampling Reveals Unknown Small Molecules
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
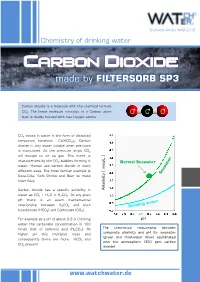
Carbon Dioxide - Made by FILTERSORB SP3
Technical Article MAR 2015 Chemistry of drinking water Carbon dioxide - made by FILTERSORB SP3 Carbon dioxide is a molecule with the chemical formula CO2. The linear molecule consists of a Carbon atom O C O that is doubly bonded with two Oxygen atoms. CO2 exists in water in the form of dissolved temporary hardness Ca(HCO3)2. Carbon dioxide is only water soluble when pressure is maintained. As the pressure drops CO2 will escape to air as gas. This event is characterized by the CO2 bubbles forming in /L ] Normal Seawater water. Human use carbon dioxide in many meq different ways. The most familiar example is Coca-Cola, Soft Drinks and Beer to make them fizzy. Carbon dioxide has a specific solubility in [ Alkalinity water as CO2 + H2O ⇌ H2CO3. At any given pH there is an exact mathematical relationship between H2CO3 and both bicarbonate (HCO3) and Carbonate (CO3). For example at a pH of about 9.3 in Drinking pH water the carbonate concentration is 100 The theoretical relationship between times that of carbonic acid (H2CO3). At carbonate alkalinity and pH for seawater higher pH this multiplier rises and (green and freshwater (blue) equilibrated consequently there are more HCO and 3 with the atmosphere (350 ppm carbon CO present 3 dioxide) www.watchwater.de Water ® Technology FILTERSORB SP3 WATCH WATER & Chemicals Making the healthiest water How SP3 Water functions in human body ? Answer: Carbon dioxide is a waste product of the around these hollow spaces will spasm and respiratory system, and of several other constrict. chemical reactions in the body such as the Transportation of oxygen to the tissues: Oxygen creation of ATP. -

A Complete Guide to Benzene
A Complete Guide to Benzene Benzene is an important organic chemical compound with the chemical formula C6H6. The benzene molecule is composed of six carbon atoms joined in a ring with one hydrogen atom attached to each. As it contains only carbon and hydrogen atoms, benzene is classed as a hydrocarbon. Benzene is a natural constituent of crude oil and is one of the elementary petrochemicals. Due to the cyclic continuous pi bond between the carbon atoms, benzene is classed as an aromatic hydrocarbon, the second [n]- annulene ([6]-annulene). It is sometimes abbreviated Ph–H. Benzene is a colourless and highly flammable liquid with a sweet smell. Source: Wikipedia Protecting people and the environment Protecting both people and the environment whilst meeting the operational needs of your business is very important and, if you have operations in the UK you will be well aware of the requirements of the CoSHH Regulations1 and likewise the Code of Federal Regulations (CFR) in the US2. Similar legislation exists worldwide, the common theme being an onus on hazard identification, risk assessment and the provision of appropriate control measures (bearing in mind the hierarchy of controls) as well as health surveillance in most cases. And whilst toxic gasses such as hydrogen sulphide and carbon monoxide are a major concern because they pose an immediate (acute) danger to life, long term exposure to relatively low level concentrations of other gasses or vapours such as volatile organic compounds (VOC) are of equal importance because of the chronic illnesses that can result from that ongoing exposure. Benzene, a common VOC Organic means the chemistry of carbon based compounds, which are substances that results from a combination of two or more different chemical elements. -

Chemical Formula
Chemical Formula Jean Brainard, Ph.D. Say Thanks to the Authors Click http://www.ck12.org/saythanks (No sign in required) AUTHOR Jean Brainard, Ph.D. To access a customizable version of this book, as well as other interactive content, visit www.ck12.org CK-12 Foundation is a non-profit organization with a mission to reduce the cost of textbook materials for the K-12 market both in the U.S. and worldwide. Using an open-content, web-based collaborative model termed the FlexBook®, CK-12 intends to pioneer the generation and distribution of high-quality educational content that will serve both as core text as well as provide an adaptive environment for learning, powered through the FlexBook Platform®. Copyright © 2013 CK-12 Foundation, www.ck12.org The names “CK-12” and “CK12” and associated logos and the terms “FlexBook®” and “FlexBook Platform®” (collectively “CK-12 Marks”) are trademarks and service marks of CK-12 Foundation and are protected by federal, state, and international laws. Any form of reproduction of this book in any format or medium, in whole or in sections must include the referral attribution link http://www.ck12.org/saythanks (placed in a visible location) in addition to the following terms. Except as otherwise noted, all CK-12 Content (including CK-12 Curriculum Material) is made available to Users in accordance with the Creative Commons Attribution-Non-Commercial 3.0 Unported (CC BY-NC 3.0) License (http://creativecommons.org/ licenses/by-nc/3.0/), as amended and updated by Creative Com- mons from time to time (the “CC License”), which is incorporated herein by this reference. -

Introduction, Formulas and Spectroscopy Overview Problem 1
Chapter 1 – Introduction, Formulas and Spectroscopy Overview Problem 1 (p 12) - Helpful equations: c = ()() and = (1 / ) so c = () / ( ) c = 3.00 x 108 m/sec = 3.00 x 1010 cm/sec a. Which photon of electromagnetic radiation below would have the longer wavelength? Convert both values to meters. -1 = 3500 cm = 4 x 1014 Hz = (1/) = (c / ) = (1/ ) = (3.0 x 108 m/s) / (4 x 1014 s-1) = (1/ cm-1)(1 m/100 cm) = 2.9 x 10-6 m = 7.5 x 10-7 m = longer wavelength = lower energy b. Which photon of electromagnetic radiation below would have the higher frequency? Convert both values to Hz. = 400 cm-1 = 300 nm = (1/ ) = 1/ [(400 cm-1)(100 cm / 1 m)] = (c/) 8 -5 9 = 2.5 x 10-5 m = (3.0 x 10 m/s) / (2.5 x 10 nm)(1m / 10 nm) v = (3.0 x 108 m/s) / (300 nm)(1m / 109 nm) = (c/) v = 1.0 x x 1015 s-1 = (3.0 x 108 m/s) / (2.5 x 10-5 m) v = 1.2 x 1013 s-1 higher frequency c. Which photon of electromagnetic radiation below would have the smaller wavenumber? Convert both values to cm-1. = 1 m = 6 x 1010 Hz = (1 / ) -1 = ( / c) = (1 / 1m) = 1 m-1 100 cm = 100 cm-1 100 cm-1 -1 = (6 x 1010 s-1 / 3.0 x 108 m/s) = 200 m-1 -1 1 m -1 = 20,000 cm smaller wavenumber 1 m Problem 2 (p 14) - Order the following photons from lowest to highest energy (first convert each value to kjoules/mole). -

H2CS) and Its Thiohydroxycarbene Isomer (HCSH
A chemical dynamics study on the gas phase formation of thioformaldehyde (H2CS) and its thiohydroxycarbene isomer (HCSH) Srinivas Doddipatlaa, Chao Hea, Ralf I. Kaisera,1, Yuheng Luoa, Rui Suna,1, Galiya R. Galimovab, Alexander M. Mebelb,1, and Tom J. Millarc,1 aDepartment of Chemistry, University of Hawai’iatManoa, Honolulu, HI 96822; bDepartment of Chemistry and Biochemistry, Florida International University, Miami, FL 33199; and cSchool of Mathematics and Physics, Queen’s University Belfast, Belfast BT7 1NN, Northern Ireland, United Kingdom Edited by Stephen J. Benkovic, The Pennsylvania State University, University Park, PA, and approved August 4, 2020 (received for review March 13, 2020) Complex organosulfur molecules are ubiquitous in interstellar molecular sulfur dioxide (SO2) (21) and sulfur (S8) (22). The second phase clouds, but their fundamental formation mechanisms have remained commences with the formation of the central protostars. Tempera- largely elusive. These processes are of critical importance in initiating a tures increase up to 300 K, and sublimation of the (sulfur-bearing) series of elementary chemical reactions, leading eventually to organo- molecules from the grains takes over (20). The subsequent gas-phase sulfur molecules—among them potential precursors to iron-sulfide chemistry exploits complex reaction networks of ion–molecule and grains and to astrobiologically important molecules, such as the amino neutral–neutral reactions (17) with models postulating that the very acid cysteine. Here, we reveal through laboratory experiments, first sulfur–carbon bonds are formed via reactions involving methyl electronic-structure theory, quasi-classical trajectory studies, and astro- radicals (CH3)andcarbene(CH2) with atomic sulfur (S) leading to chemical modeling that the organosulfur chemistry can be initiated in carbonyl monosulfide and thioformaldehyde, respectively (18). -

Ambient Water Quality Criteria for Polycyclic Aromatic Hydrocarbons (Pahs)
Ambient Water Quality Criteria For Polycyclic Aromatic Hydrocarbons (PAHs) Ministry of Environment, Lands and Parks Province of British Columbia N. K. Nagpal, Ph.D. Water Quality Branch Water Management Division February, 1993 ACKNOWLEDGEMENTS The author is indebted to the following individual and agencies for providing valuable comments during the preparation of this document. Dr. Ray Copes BC. Ministry of Health, Victoria, BC. Dr. G. R. Fox Environmental Protection Div., BC. MOELP, Victoria, BC. Mr. L. W. Pommen Water Quality Branch, BC. MOELP, Victoria, BC. Mr. R. J. Rocchini Water Quality Branch, BC. MOELP, Victoria, BC. Ms. Sherry Smith Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Mr. Scott Teed Eco-Health Branch, Conservation and Protection, Environment Canada, Hull, Quebec Ms. Bev Raymond Integrated Programs Branch, Inland Waters, Environment Canada, North Vancouver, BC. 1.0 INTRODUCTION Polycyclic aromatic hydrocarbons (PAHs) are organic compounds which are non- essential for the growth of plants, animals or humans; yet, they are ubiquitous in the environment. When present in sufficient quantity in the environment, certain PAHs are toxic and carcinogenic to plants, animals and humans. This document discusses the characteristics of PAHs and their effects on various water uses, which include drinking Ministry of Environment Water Protection and Sustainability Branch Mailing Address: Telephone: 250 387-9481 Environmental Sustainability PO Box 9362 Facsimile: 250 356-1202 and Strategic Policy Division Stn Prov Govt Website: www.gov.bc.ca/water Victoria BC V8W 9M2 water, aquatic life, wildlife, livestock watering, irrigation, recreation and aesthetics, and industrial water supplies. A significant portion of this document discusses the effects of PAHs upon aquatic life, due to its sensitivity to PAHs. -

The Life and Times of Carbon, Student Workbook
ELL Chemistry Assignment # 2 Name: __________________________________ Period: Teacher:_____________________ Collecting Carbon Lesson 1 | page 1 of 4 Where Can We Find Carbon? Carbon compounds are take up carbon dioxide from three-fourths of the dry weight everywhere around us. They the air, and use photosynthesis of plants is carbohydrates. In are in our foods, plastics, soils, to turn carbon dioxide into other words, when you look at in water, in the air, and in our carbohydrates. Carbohydrates plant matter (the stem, leaves, bodies. Carbon compounds can are carbon compounds that roots, and grains) you are come in three physical forms: store energy in the forms we call looking largely at carbohydrates. gas, liquid, and solid. Plants “starches” and “sugars.” About Animals (including humans) get their carbohydrates by eating plants, but they do not store much of what they consume. In fact, less than 1% of the body weight of animals is made up of carbohydrates. The main forms of carbon are: nonliving (abiotic) in rocks, soils, and sediments, and in water, such as bicarbonate, carbonate; living (biotic), such as plant and animal matter and dead organic matter; and carbon-based gases, such as carbon dioxide (CO2), methane (CH4), and carbon monoxide (CO). Chemists use a special system to describe compounds. They use a chemical formula of symbols that indicate the elements that make up chemical compounds. A chemical formula is also called a “molecular formula.” The chemical formula of carbon dioxide is CO2 indicating one carbon atom bonded to two oxygen atoms. The symbol for carbon is C; for hydrogen is H; for oxygen is O; for silica is Si. -

Chemical Formulas the Elements
Chemical Formulas A chemical formula gives the numbers and types of atoms that are found in a substance. When the substance is a discrete molecule, then the chemical formula is also its molecular formula. Fe (iron) is a chemical formula Fe2O3 is a molecular formula The Elements The chemical formulas of most of the elements are simply their elemental symbol: Na (sodium) Fe (iron) He (helium) U (uranium) These chemical formulas are said to be monatomic—only an atom in chemical formula 1 The Elements There are seven elements that occur naturally as diatomic molecules—molecules that contain two atoms: H2 (hydrogen) N2 (nitrogen) O2 (oxygen) F2 (fluorine) Cl2 (chlorine) Br2 (bromine) I2 (iodine) The last four elements in this list are in the same family of the Periodic Table Binary Compounds A binary compound is one composed of only two different types of atoms. Rules for binary compound formulas 1. Element to left in Periodic Table comes first except for hydrogen: KCl PCl3 Al2S3 Fe3O4 2 Binary Compounds 2. Hydrogen comes last unless other element is from group 16 or 17: LiH, NH3, B2H4, CH4 3. If both elements are from the same group, the lower element comes first: SiC, BrF3 Other Compounds For compounds with three or more elements that are not ionic, if it contains carbon, this comes first followed by hydrogen. Other elements are then listed in alphabetical order: C2H6O C4H9BrO CH3Cl C8H10N4O2 3 Other Compounds However, the preceding rule is often ignored when writing organic formulas (molecules containing carbon, hydrogen, and maybe other elements) in order to give a better idea of how the atoms are connected: C2H6O is the molecular formula for ethanol, but nobody ever writes it this way—instead the formula is written C2H5OH to indicate one H atom is connected to the O atom. -

Development of PAH Databases
Development of PAH databases Christine Joblin, Hassan Sabbah, Jean-Michel Glorian Odile Cœur-Joly, (Thierry Louge) Institut de Recherche en Astrophysique et Planétologie Université de Toulouse [UPS] – CNRS Giacomo Mulas, (Andrea Saba) INAF, Osservatorio Astronomico di Cagliari IRAP, Toulouse Seminar OVGSO Toulouse, 19/10/2018 1 Outline • The astrophysical context – The AIBs and the PAH model – I.dentification of cosmic PAHs – S.tability of cosmic PAHs – O.rigin of cosmic PAHs • The cosmic PAH experimental database – Scheme – Development of the O.rigin block • The Cagliari PAH theoretical database – Description – Implementation in VAMDC • Perspectives 2 Cosmic carbonaceous molecules Coronene (C24H12 (C60 (C2H2 Phenylalanine (C9H11NO2 3 The Aromatic Infrared Bands (AIBs) ] ] ] ] ++ + [Ar [Ar ] ] + ] ] ] ] ++ ++ [Ne [S [Ne Infrared Space Spitzer Space Observatory Telescope ESA; 1995-1998 NASA; 2003-2009 2 2 H 2 2 2 2 H 2 2 H H AKARI James Webb Space Telescope ISO SWS spectra JAXA; 2006-2011 NASA; 2019-2023? Vertratete et al. 2001, A&A 372, 981 4 PAHs in the galactic dust cycle Adapted from http://www.nrao.edu/pr/2006/gbtmolecules/ Bill Saxton, NRAO/AUI/NSF 5 The AIBs à photochemical evolution PAH clusters? VSGs NGC 7023 NW PDR PAH0 VSGs PAH0 PAH+ UV processing PAH+ 7 9 11 13 15 Wavelength (μm) Graphene? + C60 C60 Rapacioli, Joblin, Boissel, 2005, A&A 429, 193 Berné, Joblin Deville, et al. 2007, , A&A 469, 575 Berné, Mulas, Joblin, 2013, A&A 550, L4 Berné & Tielens 2012, PNAS 109, 401 6 7 8 9 10 11 Wavelength (μm) 6 I.dentification of cosmic -
Line Matching Activity
Today we use a lot of abbreviated language. Texting and instant messaging are quick ways to communicate that use as few letters and numbers as possible to get your message across. There is a shorthand method for communicating information about chemical compounds. This method uses chemical symbols and numbers to tell you which elements are in a compound and how much of each element is present. Try your hand at using this shorthand method. Complete the activity below by matching up each compound’s common name with its chemical name and chemical formula. Line Matching Activity Instructions Match the following chemical formulas with their chemical names: Chemical Formula Chemical Name Co cobalt chloride H2O baking soda CO2 sulfuric acid NaHCO3 water Br2 cobalt H2SO4 carbon dioxide CoCl2 bromine Chemical names can be very long. Fortunately, we have an abbreviated way to communicate them using symbols and numbers. Chemical formulas have two parts: the symbol of each element in a molecule of the substance a number indicating how many atoms of each element are in each molecule of the substance We will start with a simple example. The chemical formula for the oxygen gas we breathe is O2. There is only one element in oxygen gas: oxygen. O is the symbol for the element oxygen, and the subscript 2 means that each molecule of oxygen contains two atoms of oxygen. The atoms of oxygen are bonded together covalently by sharing pairs of electrons. SubscriptsPrefixes © 2009 Jupiterimages Corporation The same principle applies to molecules of compounds, which contain atoms of more than one element. -

Experimental Evidence for Glycolaldehyde and Ethylene Glycol Formation by Surface Hydrogenation of CO Molecules Under Dense Molecular Cloud Conditions
Experimental evidence for Glycolaldehyde and Ethylene Glycol formation by surface hydrogenation of CO molecules under dense molecular cloud conditions G. Fedoseev1, a), H. M. Cuppen2, S. Ioppolo2, T. Lamberts1, 2 and H. Linnartz1 1Sackler Laboratory for Astrophysics, Leiden Observatory, University of Leiden, PO Box 9513, NL 2300 RA Leiden, The Netherlands 2Institute for Molecules and Materials, Radboud University Nijmegen, PO Box 9010, NL 6500 GL Nijmegen, The Netherlands Abstract This study focuses on the formation of two molecules of astrobiological importance - glycolaldehyde (HC(O)CH2OH) and ethylene glycol (H2C(OH)CH2OH) - by surface hydrogenation of CO molecules. Our experiments aim at simulating the CO freeze-out stage in interstellar dark cloud regions, well before thermal and energetic processing become dominant. It is shown that along with the formation of H2CO and CH3OH – two well established products of CO hydrogenation – also molecules with more than one carbon atom form. The key step in this process is believed to be the recombination of two HCO radicals followed by the formation of a C-C bond. The experimentally established reaction pathways are implemented into a continuous-time random-walk Monte Carlo model, previously used to model the formation of CH3OH on astrochemical time-scales, to study their impact on the solid-state abundances in dense interstellar clouds of glycolaldehyde and ethylene glycol. Key words: astrochemistry – methods: laboratory – ISM: atoms – ISM: molecules – infrared: ISM. 1 Introduction Among approximately 180 molecules identified in the inter- and circumstellar medium over 50 molecules comprise of six or more atoms. For astrochemical standards, these are seen as ‘complex’ species. -

671-2121 Vinegar Molecule Vinegar Is a Naturally-Occurring Liquid That Contains Many Chemicals
©2016 - v 6/16 671-2121 Vinegar Molecule Vinegar is a naturally-occurring liquid that contains many chemicals. It is approximately 5% acetic acid in wa- ter. The molecular formula for water is H2O. The structural formula for acetic acid is CH3COOH. Acetic acid /əˈsiːtᵻk/, systematically named ethanoic acid /ˌɛθəˈnoʊᵻk/, is a colourless liquid organic compound with the chemical formula CH3COOH (also written as CH3CO2H or C2H4O2). When undiluted, it is sometimes called glacial acetic acid. Vinegar is roughly 3–9% acetic acid by volume, making acetic acid the main com- ponent of vinegar apart from water. Acetic acid has a distinctive sour taste and pungent smell. In addition to household vinegar, it is mainly produced as a precursor to polyvinyl acetate and cellulose acetate. Although it is classified as a weak acid, concentrated acetic acid is corrosive and can attack the skin. To easily assemble your molecule, notice the holes on each particular atom. Press a connector firmly into this hole until it is flush with the surface of the atom. The connection should be firm but still easy to disassemble. Your molecule can be put together and taken apart as many times as you wish. Warranty and Parts: We replace all defective or missing parts free of charge. Additional replacement parts may be ordered toll-free. We accept MasterCard, Visa, checks and School P.O.s. All products warranted to be free from defect for 90 days. Does not apply to accident, misuse or normal wear and tear. Intended for children 13 years of age and up. This item is not a toy.