The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intelligent Design, Abiogenesis, and Learning from History: Dennis R
Author Exchange Intelligent Design, Abiogenesis, and Learning from History: Dennis R. Venema A Reply to Meyer Dennis R. Venema Weizsäcker’s book The World View of Physics is still keeping me very busy. It has again brought home to me quite clearly how wrong it is to use God as a stop-gap for the incompleteness of our knowledge. If in fact the frontiers of knowledge are being pushed back (and that is bound to be the case), then God is being pushed back with them, and is therefore continually in retreat. We are to find God in what we know, not in what we don’t know; God wants us to realize his presence, not in unsolved problems but in those that are solved. Dietrich Bonhoeffer1 am thankful for this opportunity to nature, is the result of intelligence. More- reply to Stephen Meyer’s criticisms over, this assertion is proffered as the I 2 of my review of his book Signature logical basis for inferring design for the in the Cell (hereafter Signature). Meyer’s origin of biological information: if infor- critiques of my review fall into two gen- mation only ever arises from intelli- eral categories. First, he claims I mistook gence, then the mere presence of Signature for an argument against bio- information demonstrates design. A few logical evolution, rendering several of examples from Signature make the point my arguments superfluous. Secondly, easily: Meyer asserts that I have failed to refute … historical scientists can show that his thesis by not providing a “causally a presently acting cause must have adequate alternative explanation” for the been present in the past because the origin of life in that the few relevant cri- proposed candidate is the only known tiques I do provide are “deeply flawed.” cause of the effect in question. -

Revised Glossary for AQA GCSE Biology Student Book
Biology Glossary amino acids small molecules from which proteins are A built abiotic factor physical or non-living conditions amylase a digestive enzyme (carbohydrase) that that affect the distribution of a population in an breaks down starch ecosystem, such as light, temperature, soil pH anaerobic respiration respiration without using absorption the process by which soluble products oxygen of digestion move into the blood from the small intestine antibacterial chemicals chemicals produced by plants as a defence mechanism; the amount abstinence method of contraception whereby the produced will increase if the plant is under attack couple refrains from intercourse, particularly when an egg might be in the oviduct antibiotic e.g. penicillin; medicines that work inside the body to kill bacterial pathogens accommodation ability of the eyes to change focus antibody protein normally present in the body acid rain rain water which is made more acidic by or produced in response to an antigen, which it pollutant gases neutralises, thus producing an immune response active site the place on an enzyme where the antimicrobial resistance (AMR) an increasing substrate molecule binds problem in the twenty-first century whereby active transport in active transport, cells use energy bacteria have evolved to develop resistance against to transport substances through cell membranes antibiotics due to their overuse against a concentration gradient antiretroviral drugs drugs used to treat HIV adaptation features that organisms have to help infections; they -

Impacts of a Comprehensive Public Engagement Training and Support Program on Scientists’ Outreach Attitudes and Practices
International Journal of Science Education, Part B Communication and Public Engagement ISSN: 2154-8455 (Print) 2154-8463 (Online) Journal homepage: http://www.tandfonline.com/loi/rsed20 Impacts of a comprehensive public engagement training and support program on scientists’ outreach attitudes and practices Cathlyn Stylinski, Martin Storksdieck, Nicolette Canzoneri, Eve Klein & Anna Johnson To cite this article: Cathlyn Stylinski, Martin Storksdieck, Nicolette Canzoneri, Eve Klein & Anna Johnson (2018): Impacts of a comprehensive public engagement training and support program on scientists’ outreach attitudes and practices, International Journal of Science Education, Part B, DOI: 10.1080/21548455.2018.1506188 To link to this article: https://doi.org/10.1080/21548455.2018.1506188 Published online: 31 Aug 2018. Submit your article to this journal View Crossmark data Full Terms & Conditions of access and use can be found at http://www.tandfonline.com/action/journalInformation?journalCode=rsed20 INTERNATIONAL JOURNAL OF SCIENCE EDUCATION, PART B https://doi.org/10.1080/21548455.2018.1506188 Impacts of a comprehensive public engagement training and support program on scientists’ outreach attitudes and practices Cathlyn Stylinski a, Martin Storksdieck b, Nicolette Canzonerib, Eve Kleinc and Anna Johnson c aUMCES, University of Maryland Center for Environmental Science Appalachian Laboratory, Frostburg, MD, USA; bCenter for Research on Lifelong STEM Learning, Oregon State University, Corvallis, OR, USA; cInstitute for Learning Innovation, Seattle, WA, USA ABSTRACT ARTICLE HISTORY Scientists are increasingly being called upon to play a more prominent role Received 1 November 2017 in the interface of science and society by contributing to science literacy in Accepted 24 July 2018 ways that support two-way exchanges with the public. -

THE INDY STEM TEACHER RESIDENCY PROGRAM: a Teacher Quality Partnership Between Indianapolis Public Schools and Purdue University
Table of Contents QUALITY OF THE PROJECT DESIGN 1-29 1.Extent to which the proposed project demonstrates a rationale 3-7 (a) Needs Assessment data 4-6 AppC (b) Project will prepare teachers with strong teaching skills 1-29 2.Goals, objectives, and outcomes are clearly specified and measurable 42-44 3.Project is designed to build capacity / yield results that extend beyond project period 29 4.Project represents exceptional approach for meeting statutory purposes/requirements 1-29 (c) Program will prepare teachers to understand/use research data to improve instruction 1-29 (f5i) Program aligns with state early learning standards… 1-29 ADEQUACY OF RESOURCES 29-33 1. Adequacy of support, including facilities, equipment, supplies, and other resources 29-33 (e1) Assessment describing resources, including integration of funds from other sources 29-33 (e2) Assessment that describes the intended use of the grant funds 33 2. Relevance/demonstrated commitment of partners to project implementation/success 33 (e3) Commitment of resources and to the continuation of activities when the grant ends 29-33 QUALITY OF MANAGEMENT PLAN 33-41 1. Adequacy of management plan to achieve objectives on time/within budget, 33-39, including clearly defined responsibilities, timelines, and milestones for AppJ accomplishing tasks (d1) Coordination of strategies/activities with funded teacher preparation or PD prog 13 (d2) How activities are consistent with State, local, and other education reform activities 8, 18- 19,21- that promote teacher quality and student academic -

Opportunities for Catalysis in the 21St Century
Opportunities for Catalysis in The 21st Century A Report from the Basic Energy Sciences Advisory Committee BASIC ENERGY SCIENCES ADVISORY COMMITTEE SUBPANEL WORKSHOP REPORT Opportunities for Catalysis in the 21st Century May 14-16, 2002 Workshop Chair Professor J. M. White University of Texas Writing Group Chair Professor John Bercaw California Institute of Technology This page is intentionally left blank. Contents Executive Summary........................................................................................... v A Grand Challenge....................................................................................................... v The Present Opportunity .............................................................................................. v The Importance of Catalysis Science to DOE.............................................................. vi A Recommendation for Increased Federal Investment in Catalysis Research............. vi I. Introduction................................................................................................ 1 A. Background, Structure, and Organization of the Workshop .................................. 1 B. Recent Advances in Experimental and Theoretical Methods ................................ 1 C. The Grand Challenge ............................................................................................. 2 D. Enabling Approaches for Progress in Catalysis ..................................................... 3 E. Consensus Observations and Recommendations.................................................. -
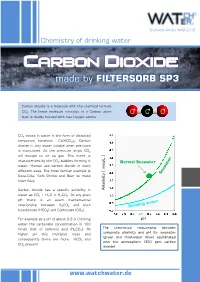
Carbon Dioxide - Made by FILTERSORB SP3
Technical Article MAR 2015 Chemistry of drinking water Carbon dioxide - made by FILTERSORB SP3 Carbon dioxide is a molecule with the chemical formula CO2. The linear molecule consists of a Carbon atom O C O that is doubly bonded with two Oxygen atoms. CO2 exists in water in the form of dissolved temporary hardness Ca(HCO3)2. Carbon dioxide is only water soluble when pressure is maintained. As the pressure drops CO2 will escape to air as gas. This event is characterized by the CO2 bubbles forming in /L ] Normal Seawater water. Human use carbon dioxide in many meq different ways. The most familiar example is Coca-Cola, Soft Drinks and Beer to make them fizzy. Carbon dioxide has a specific solubility in [ Alkalinity water as CO2 + H2O ⇌ H2CO3. At any given pH there is an exact mathematical relationship between H2CO3 and both bicarbonate (HCO3) and Carbonate (CO3). For example at a pH of about 9.3 in Drinking pH water the carbonate concentration is 100 The theoretical relationship between times that of carbonic acid (H2CO3). At carbonate alkalinity and pH for seawater higher pH this multiplier rises and (green and freshwater (blue) equilibrated consequently there are more HCO and 3 with the atmosphere (350 ppm carbon CO present 3 dioxide) www.watchwater.de Water ® Technology FILTERSORB SP3 WATCH WATER & Chemicals Making the healthiest water How SP3 Water functions in human body ? Answer: Carbon dioxide is a waste product of the around these hollow spaces will spasm and respiratory system, and of several other constrict. chemical reactions in the body such as the Transportation of oxygen to the tissues: Oxygen creation of ATP. -

Astro2020 Science White Paper Unlocking the Secrets of Late-Stage Stellar Evolution and Mass Loss Through Radio Wavelength Imaging
Astro2020 Science White Paper Unlocking the Secrets of Late-Stage Stellar Evolution and Mass Loss through Radio Wavelength Imaging Thematic Areas: Planetary Systems Star and Planet Formation Formation and Evolution of Compact Objects Cosmology and Fundamental Physics 7 Stars and Stellar Evolution Resolved Stellar Populations and their Environments Galaxy Evolution Multi-Messenger Astronomy and Astrophysics Principal Author: Name: Lynn D. Matthews Institution: Massachusetts Institute of Technology Haystack Observatory Email: [email protected] Co-authors: Mark J Claussen (National Radio Astronomy Observatory) Graham M. Harper (University of Colorado - Boulder) Karl M. Menten (Max Planck Institut fur¨ Radioastronomie) Stephen Ridgway (National Optical Astronomy Observatory) Executive Summary: During the late phases of evolution, low-to-intermediate mass stars like our Sun undergo periods of extensive mass loss, returning up to 80% of their initial mass to the interstellar medium. This mass loss profoundly affects the stellar evolutionary history, and the resulting circumstellar ejecta are a primary source of dust and heavy element enrichment in the Galaxy. However, many details concerning the physics of late-stage stellar mass loss remain poorly understood, including the wind launching mechanism(s), the mass loss geometry and timescales, and the mass loss histories of stars of various initial masses. These uncertainties have implications not only for stellar astrophysics, but for fields ranging from star formation to extragalactic astronomy and cosmology. Observations at centimeter, millimeter, and submillimeter wavelengths that resolve the radio surfaces and extended atmospheres of evolved arXiv:1903.05592v1 [astro-ph.SR] 13 Mar 2019 stars in space, time, and frequency are poised to provide groundbreaking new insights into these questions in the coming decade. -

Bayesian Analysis of the Astrobiological Implications of Life's
Bayesian analysis of the astrobiological implications of life's early emergence on Earth David S. Spiegel ∗ y, Edwin L. Turner y z ∗Institute for Advanced Study, Princeton, NJ 08540,yDept. of Astrophysical Sciences, Princeton Univ., Princeton, NJ 08544, USA, and zInstitute for the Physics and Mathematics of the Universe, The Univ. of Tokyo, Kashiwa 227-8568, Japan Submitted to Proceedings of the National Academy of Sciences of the United States of America Life arose on Earth sometime in the first few hundred million years Any inferences about the probability of life arising (given after the young planet had cooled to the point that it could support the conditions present on the early Earth) must be informed water-based organisms on its surface. The early emergence of life by how long it took for the first living creatures to evolve. By on Earth has been taken as evidence that the probability of abiogen- definition, improbable events generally happen infrequently. esis is high, if starting from young-Earth-like conditions. We revisit It follows that the duration between events provides a metric this argument quantitatively in a Bayesian statistical framework. By (however imperfect) of the probability or rate of the events. constructing a simple model of the probability of abiogenesis, we calculate a Bayesian estimate of its posterior probability, given the The time-span between when Earth achieved pre-biotic condi- data that life emerged fairly early in Earth's history and that, billions tions suitable for abiogenesis plus generally habitable climatic of years later, curious creatures noted this fact and considered its conditions [5, 6, 7] and when life first arose, therefore, seems implications. -

Human Anatomy (Biology 2) Lecture Notes Updated July 2017 Instructor
Human Anatomy (Biology 2) Lecture Notes Updated July 2017 Instructor: Rebecca Bailey 1 Chapter 1 The Human Body: An Orientation • Terms - Anatomy: the study of body structure and relationships among structures - Physiology: the study of body function • Levels of Organization - Chemical level 1. atoms and molecules - Cells 1. the basic unit of all living things - Tissues 1. cells join together to perform a particular function - Organs 1. tissues join together to perform a particular function - Organ system 1. organs join together to perform a particular function - Organismal 1. the whole body • Organ Systems • Anatomical Position • Regional Names - Axial region 1. head 2. neck 3. trunk a. thorax b. abdomen c. pelvis d. perineum - Appendicular region 1. limbs • Directional Terms - Superior (above) vs. Inferior (below) - Anterior (toward the front) vs. Posterior (toward the back)(Dorsal vs. Ventral) - Medial (toward the midline) vs. Lateral (away from the midline) - Intermediate (between a more medial and a more lateral structure) - Proximal (closer to the point of origin) vs. Distal (farther from the point of origin) - Superficial (toward the surface) vs. Deep (away from the surface) • Planes and Sections divide the body or organ - Frontal or coronal 1. divides into anterior/posterior 2 - Sagittal 1. divides into right and left halves 2. includes midsagittal and parasagittal - Transverse or cross-sectional 1. divides into superior/inferior • Body Cavities - Dorsal 1. cranial cavity 2. vertebral cavity - Ventral 1. lined with serous membrane 2. viscera (organs) covered by serous membrane 3. thoracic cavity a. two pleural cavities contain the lungs b. pericardial cavity contains heart c. the cavities are defined by serous membrane d. -

Lecture 2 Our Place in the Universe (Cont'd)
Astronomy 110 –1 Lecture 2 Our Place in the Universe (Cont’d) 14/01/09 1 Copyright © 2009 Pearson Education, Inc. A few useful mathematical skills 14/01/09 2 Copyright © 2009 Pearson Education, Inc. Powers of ten 103 = 10 x 10 x 10 = 1000 102 = 10 x 10 = 100 101 = 10 100 = 1 10-1 = 1/10 = 0.1 10-2 = 1/10 x 1/10 = 0.01 10-3 = 1/10 x 1/10 x 1/10 = 0.001 Then: 300 = 3 x 100 = 3 x 102 2,500 = 2.5 x 1000 = 2.5 x 103 14/01/09 3 Copyright © 2009 Pearson Education, Inc. Multiplying & dividing 101 x 101 = 100 = 102 101 x 102 = 10 x 100 = 1000 = 103 102 x 102 = 100 x 100 = 10000 = 104 When multiplying two powers of ten, add the exponents 1000 ÷ 100 = 103 ÷ 102 = 10 = 101 10 ÷ 10 = 101 ÷ 101 = 1 = 100 10 ÷ 100 = 101 ÷ 102 = 1/10 = 10-1 When dividing, subtract exponent of divisor from exponent of numerator 14/01/09 4 Copyright © 2009 Pearson Education, Inc. Powers and roots (104)3 = 104 x 104 x 104 = 1012 Rule of thumb: when raising to a power, multiply exponents What about √(104)? √(104) = 102 taking roots is the same as raising to a fractional power, in this case 1/2 power: √(104) = (104)1/2 = 102 √ is the same as raising to 1/2 power 3√ is the same as raising to 1/3 power 4√ is the same as raising to 1/4 power 14/01/09 5 Copyright © 2009 Pearson Education, Inc. -

Dehydrogenation by Heterogeneous Catalysts
Dehydrogenation by Heterogeneous Catalysts Daniel E. Resasco School of Chemical Engineering and Materials Science University of Oklahoma Encyclopedia of Catalysis January, 2000 1. INTRODUCTION Catalytic dehydrogenation of alkanes is an endothermic reaction, which occurs with an increase in the number of moles and can be represented by the expression Alkane ! Olefin + Hydrogen This reaction cannot be carried out thermally because it is highly unfavorable compared to the cracking of the hydrocarbon, since the C-C bond strength (about 246 kJ/mol) is much lower than that of the C-H bond (about 363 kJ/mol). However, in the presence of a suitable catalyst, dehydrogenation can be carried out with minimal C-C bond rupture. The strong C-H bond is a closed-shell σ orbital that can be activated by oxide or metal catalysts. Oxides can activate the C-H bond via hydrogen abstraction because they can form O-H bonds, which can have strengths comparable to that of the C- H bond. By contrast, metals cannot accomplish the hydrogen abstraction because the M- H bonds are much weaker than the C-H bond. However, the sum of the M-H and M-C bond strengths can exceed the C-H bond strength, making the process thermodynamically possible. In this case, the reaction is thought to proceed via a three centered transition state, which can be described as a metal atom inserting into the C-H bond. The C-H bond bridges across the metal atom until it breaks, followed by the formation of the corresponding M-H and M-C bonds.1 Therefore, dehydrogenation of alkanes can be carried out on oxides as well as on metal catalysts. -

A Complete Guide to Benzene
A Complete Guide to Benzene Benzene is an important organic chemical compound with the chemical formula C6H6. The benzene molecule is composed of six carbon atoms joined in a ring with one hydrogen atom attached to each. As it contains only carbon and hydrogen atoms, benzene is classed as a hydrocarbon. Benzene is a natural constituent of crude oil and is one of the elementary petrochemicals. Due to the cyclic continuous pi bond between the carbon atoms, benzene is classed as an aromatic hydrocarbon, the second [n]- annulene ([6]-annulene). It is sometimes abbreviated Ph–H. Benzene is a colourless and highly flammable liquid with a sweet smell. Source: Wikipedia Protecting people and the environment Protecting both people and the environment whilst meeting the operational needs of your business is very important and, if you have operations in the UK you will be well aware of the requirements of the CoSHH Regulations1 and likewise the Code of Federal Regulations (CFR) in the US2. Similar legislation exists worldwide, the common theme being an onus on hazard identification, risk assessment and the provision of appropriate control measures (bearing in mind the hierarchy of controls) as well as health surveillance in most cases. And whilst toxic gasses such as hydrogen sulphide and carbon monoxide are a major concern because they pose an immediate (acute) danger to life, long term exposure to relatively low level concentrations of other gasses or vapours such as volatile organic compounds (VOC) are of equal importance because of the chronic illnesses that can result from that ongoing exposure. Benzene, a common VOC Organic means the chemistry of carbon based compounds, which are substances that results from a combination of two or more different chemical elements.