The Dissipative Photochemical Origin of Life: UVC Abiogenesis of Adenine
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Glossary - Cellbiology
1 Glossary - Cellbiology Blotting: (Blot Analysis) Widely used biochemical technique for detecting the presence of specific macromolecules (proteins, mRNAs, or DNA sequences) in a mixture. A sample first is separated on an agarose or polyacrylamide gel usually under denaturing conditions; the separated components are transferred (blotting) to a nitrocellulose sheet, which is exposed to a radiolabeled molecule that specifically binds to the macromolecule of interest, and then subjected to autoradiography. Northern B.: mRNAs are detected with a complementary DNA; Southern B.: DNA restriction fragments are detected with complementary nucleotide sequences; Western B.: Proteins are detected by specific antibodies. Cell: The fundamental unit of living organisms. Cells are bounded by a lipid-containing plasma membrane, containing the central nucleus, and the cytoplasm. Cells are generally capable of independent reproduction. More complex cells like Eukaryotes have various compartments (organelles) where special tasks essential for the survival of the cell take place. Cytoplasm: Viscous contents of a cell that are contained within the plasma membrane but, in eukaryotic cells, outside the nucleus. The part of the cytoplasm not contained in any organelle is called the Cytosol. Cytoskeleton: (Gk. ) Three dimensional network of fibrous elements, allowing precisely regulated movements of cell parts, transport organelles, and help to maintain a cell’s shape. • Actin filament: (Microfilaments) Ubiquitous eukaryotic cytoskeletal proteins (one end is attached to the cell-cortex) of two “twisted“ actin monomers; are important in the structural support and movement of cells. Each actin filament (F-actin) consists of two strands of globular subunits (G-Actin) wrapped around each other to form a polarized unit (high ionic cytoplasm lead to the formation of AF, whereas low ion-concentration disassembles AF). -

Dissipative Mechanics Using Complex-Valued Hamiltonians
Dissipative Mechanics Using Complex-Valued Hamiltonians S. G. Rajeev Department of Physics and Astronomy University of Rochester, Rochester, New York 14627 March 18, 2018 Abstract We show that a large class of dissipative systems can be brought to a canonical form by introducing complex co-ordinates in phase space and a complex-valued hamiltonian. A naive canonical quantization of these sys- tems lead to non-hermitean hamiltonian operators. The excited states are arXiv:quant-ph/0701141v1 19 Jan 2007 unstable and decay to the ground state . We also compute the tunneling amplitude across a potential barrier. 1 1 Introduction In many physical situations, loss of energy of the system under study to the out- side environment cannot be ignored. Often, the long time behavior of the system is determined by this loss of energy, leading to interesting phenomena such as attractors. There is an extensive literature on dissipative systems at both the classical and quantum levels (See for example the textbooks [1, 2, 3]). Often the theory is based on an evolution equation of the density matrix of a ‘small system’ coupled to a ‘reservoir’ with a large number of degrees of freedom, after the reservoir has been averaged out. In such approaches the system is described by a mixed state rather than a pure state: in quantum mechanics by a density instead of a wavefunction and in classical mechanics by a density function rather than a point in the phase space. There are other approaches that do deal with the evolution equations of a pure state. The canonical formulation of classical mechanics does not apply in a direct way to dissipative systems because the hamiltonian usually has the meaning of energy and would be conserved. -

Quantum Biology: an Update and Perspective
quantum reports Review Quantum Biology: An Update and Perspective Youngchan Kim 1,2,3 , Federico Bertagna 1,4, Edeline M. D’Souza 1,2, Derren J. Heyes 5 , Linus O. Johannissen 5 , Eveliny T. Nery 1,2 , Antonio Pantelias 1,2 , Alejandro Sanchez-Pedreño Jimenez 1,2 , Louie Slocombe 1,6 , Michael G. Spencer 1,3 , Jim Al-Khalili 1,6 , Gregory S. Engel 7 , Sam Hay 5 , Suzanne M. Hingley-Wilson 2, Kamalan Jeevaratnam 4, Alex R. Jones 8 , Daniel R. Kattnig 9 , Rebecca Lewis 4 , Marco Sacchi 10 , Nigel S. Scrutton 5 , S. Ravi P. Silva 3 and Johnjoe McFadden 1,2,* 1 Leverhulme Quantum Biology Doctoral Training Centre, University of Surrey, Guildford GU2 7XH, UK; [email protected] (Y.K.); [email protected] (F.B.); e.d’[email protected] (E.M.D.); [email protected] (E.T.N.); [email protected] (A.P.); [email protected] (A.S.-P.J.); [email protected] (L.S.); [email protected] (M.G.S.); [email protected] (J.A.-K.) 2 Department of Microbial and Cellular Sciences, School of Bioscience and Medicine, Faculty of Health and Medical Sciences, University of Surrey, Guildford GU2 7XH, UK; [email protected] 3 Advanced Technology Institute, University of Surrey, Guildford GU2 7XH, UK; [email protected] 4 School of Veterinary Medicine, Faculty of Health and Medical Sciences, University of Surrey, Guildford GU2 7XH, UK; [email protected] (K.J.); [email protected] (R.L.) 5 Manchester Institute of Biotechnology, Department of Chemistry, The University of Manchester, -

Donor-Acceptor Interaction and Photochemistry of Polymethylene-Linked Bichromophores in Solution
9042 J. Am. Chem. Soc. 1996, 118, 9042-9051 Donor-Acceptor Interaction and Photochemistry of Polymethylene-Linked Bichromophores in Solution Song-lei Zhang, Matthew J. Lang, Steven Goodman,† Christopher Durnell, Vlastimil Fidlar,‡ Graham R. Fleming, and Nien-chu C. Yang* Contribution from the Department of Chemistry, UniVersity of Chicago, Chicago, Illinois 60637 ReceiVed July 17, 1996X Abstract: The ground-state and the excited-state spectroscopic properties of four series of polymethylene-linked anthracene-dialkylaniline bichromophores were compared as a probe to the relationship between energetics and distance in photoinduced electron transfer (PET). The results demonstrate that, when the energy level of the charge- transfer (CT) state is lowered below that of the localized excited state by appropriate substituents, there is a strong electron-donor-acceptor (EDA) interaction in the ground state which is absent in other bichromophores. Absorption and fluorescence excitation studies revealed that there is an unusually strong EDA interaction in the ground state of A-2 which is absent in other members in the A series. When A-2 is excited directly into this EDA absorption, it exhibits two CT emissions, one at 490 nm and the other at 605 nm. The quantum yield (τf) and the lifetime (Φf) of the two emissions are dependent on the viscosity of the alkane solvent. The Φf and the τf of the 490 nm emission increased when the solvent viscosity was increased; however, those of the 605 nm emission remained essentially unchanged. The risetime of the 605 nm emission is 420 ps, but that of the 490 nm emission is instrument-function limiting. -

An Ab Initio Definition of Life Pertaining to Astrobiology Ian Von Hegner
An ab initio definition of life pertaining to Astrobiology Ian von Hegner To cite this version: Ian von Hegner. An ab initio definition of life pertaining to Astrobiology. 2019. hal-02272413v2 HAL Id: hal-02272413 https://hal.archives-ouvertes.fr/hal-02272413v2 Preprint submitted on 15 Oct 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. HAL archives-ouvertes.fr | CCSD, August, 2019. An ab initio definition of life pertaining to Astrobiology Ian von Hegner Aarhus University Abstract Many definitions of life have been put forward in the course of time, but none have emerged to entirely encapsulate life. Putting forward an adequate definition is not a simple matter to do, despite many people seeming to believe they have an intuitive understanding of what is meant when it is stated that something is life. However, it is important to define life, because we ourselves, individually and collectively, are life, which entails an importance in itself. Furthermore, humankind‘s capability to look for life on other planets is steadily becoming a real possibility. But in order to realize that search, a definition of life is required. -

Quantum Crystallography†
Chemical Science View Article Online MINIREVIEW View Journal | View Issue Quantum crystallography† Simon Grabowsky,*a Alessandro Genoni*bc and Hans-Beat Burgi*de Cite this: Chem. Sci.,2017,8,4159 ¨ Approximate wavefunctions can be improved by constraining them to reproduce observations derived from diffraction and scattering experiments. Conversely, charge density models, incorporating electron-density distributions, atomic positions and atomic motion, can be improved by supplementing diffraction Received 16th December 2016 experiments with quantum chemically calculated, tailor-made electron densities (form factors). In both Accepted 3rd March 2017 cases quantum chemistry and diffraction/scattering experiments are combined into a single, integrated DOI: 10.1039/c6sc05504d tool. The development of quantum crystallographic research is reviewed. Some results obtained by rsc.li/chemical-science quantum crystallography illustrate the potential and limitations of this field. 1. Introduction today's organic, inorganic and physical chemistry. These tools are usually employed separately. Diffraction and scattering Quantum chemistry methods and crystal structure determina- experiments provide structures at the atomic scale, while the Creative Commons Attribution 3.0 Unported Licence. tion are highly developed research tools, indispensable in techniques of quantum chemistry provide wavefunctions and aUniversitat¨ Bremen, Fachbereich 2 – Biologie/Chemie, Institut fur¨ Anorganische eUniversitat¨ Zurich,¨ Institut fur¨ Chemie, Winterthurerstrasse 190, CH-8057 Zurich,¨ Chemie und Kristallographie, Leobener Str. NW2, 28359 Bremen, Germany. Switzerland E-mail: [email protected] † Dedicated to Prof. Louis J. Massa on the occasion of his 75th birthday and to bCNRS, Laboratoire SRSMC, UMR 7565, Vandoeuvre-l`es-Nancy, F-54506, France Prof. Dylan Jayatilaka on the occasion of his 50th birthday in recognition of their cUniversit´e de Lorraine, Laboratoire SRSMC, UMR 7565, Vandoeuvre-l`es-Nancy, F- pioneering contributions to the eld of X-ray quantum crystallography. -
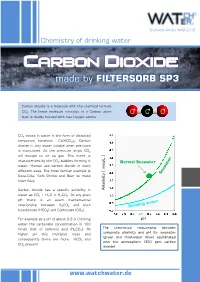
Carbon Dioxide - Made by FILTERSORB SP3
Technical Article MAR 2015 Chemistry of drinking water Carbon dioxide - made by FILTERSORB SP3 Carbon dioxide is a molecule with the chemical formula CO2. The linear molecule consists of a Carbon atom O C O that is doubly bonded with two Oxygen atoms. CO2 exists in water in the form of dissolved temporary hardness Ca(HCO3)2. Carbon dioxide is only water soluble when pressure is maintained. As the pressure drops CO2 will escape to air as gas. This event is characterized by the CO2 bubbles forming in /L ] Normal Seawater water. Human use carbon dioxide in many meq different ways. The most familiar example is Coca-Cola, Soft Drinks and Beer to make them fizzy. Carbon dioxide has a specific solubility in [ Alkalinity water as CO2 + H2O ⇌ H2CO3. At any given pH there is an exact mathematical relationship between H2CO3 and both bicarbonate (HCO3) and Carbonate (CO3). For example at a pH of about 9.3 in Drinking pH water the carbonate concentration is 100 The theoretical relationship between times that of carbonic acid (H2CO3). At carbonate alkalinity and pH for seawater higher pH this multiplier rises and (green and freshwater (blue) equilibrated consequently there are more HCO and 3 with the atmosphere (350 ppm carbon CO present 3 dioxide) www.watchwater.de Water ® Technology FILTERSORB SP3 WATCH WATER & Chemicals Making the healthiest water How SP3 Water functions in human body ? Answer: Carbon dioxide is a waste product of the around these hollow spaces will spasm and respiratory system, and of several other constrict. chemical reactions in the body such as the Transportation of oxygen to the tissues: Oxygen creation of ATP. -

Atomic and Molecular Laser-Induced Breakdown Spectroscopy of Selected Pharmaceuticals
Article Atomic and Molecular Laser-Induced Breakdown Spectroscopy of Selected Pharmaceuticals Pravin Kumar Tiwari 1,2, Nilesh Kumar Rai 3, Rohit Kumar 3, Christian G. Parigger 4 and Awadhesh Kumar Rai 2,* 1 Institute for Plasma Research, Gandhinagar, Gujarat-382428, India 2 Laser Spectroscopy Research Laboratory, Department of Physics, University of Allahabad, Prayagraj-211002, India 3 CMP Degree College, Department of Physics, University of Allahabad, Pragyagraj-211002, India 4 Physics and Astronomy Department, University of Tennessee, University of Tennessee Space Institute, Center for Laser Applications, 411 B.H. Goethert Parkway, Tullahoma, TN 37388-9700, USA * Correspondence: [email protected]; Tel.: +91-532-2460993 Received: 10 June 2019; Accepted: 10 July 2019; Published: 19 July 2019 Abstract: Laser-induced breakdown spectroscopy (LIBS) of pharmaceutical drugs that contain paracetamol was investigated in air and argon atmospheres. The characteristic neutral and ionic spectral lines of various elements and molecular signatures of CN violet and C2 Swan band systems were observed. The relative hardness of all drug samples was measured as well. Principal component analysis, a multivariate method, was applied in the data analysis for demarcation purposes of the drug samples. The CN violet and C2 Swan spectral radiances were investigated for evaluation of a possible correlation of the chemical and molecular structures of the pharmaceuticals. Complementary Raman and Fourier-transform-infrared spectroscopies were used to record the molecular spectra of the drug samples. The application of the above techniques for drug screening are important for the identification and mitigation of drugs that contain additives that may cause adverse side-effects. Keywords: paracetamol; laser-induced breakdown spectroscopy; cyanide; carbon swan bands; principal component analysis; Raman spectroscopy; Fourier-transform-infrared spectroscopy 1. -

Quantum Mechanics and Biology
QUANTUM MECHANICS AND BIOLOGY H. C. LONGUET-HIGGINS From the University of Cambridge, England I should like to begin by saying that I feel very diffident about speaking on the subject which the organizers of this Conference invited me to discuss with you. What I have to say will inevitably be limited by the extreme superficiality of my acquaintance with modem biochemistry and biophysics. The only apology which I can offer for being here at all is that I feel, in common with most scientists, that it is a good thing for workers in quite different fields to meet occasionally and compare notes so as to discover whether the methods of one discipline can usefully be applied to the problems of another. It will soon become apparent to you that my opinions on the usefulness of quantum mechanics to biologists are decidedly con- servative; but if these opinions are not generally shared, perhaps at least they will provoke discussion. The thesis which I want to develop in the next few minutes will seem to many of you to be rather pedestrian. Briefly I want to suggest that for many years to come the student of living matter will have much more need for an understanding of physical chemistry than for a knowledge of quantum mechanics. However much one may marvel at the variety and versatility of living things, it cannot be denied that all organisms are in one sense physico-chemical machines. Ingenious and baffling as their structure and function may seem to be, it is therefore proper, in attempting to understand them, to compare their component parts with those in- animate systems which the physical chemist now understands more or less thor- oughly. -

Dissipative Structures in Educational Change: Prigogine and the Academy
Wichita State University Libraries SOAR: Shocker Open Access Repository Donald L. Gilstrap University Libraries Dissipative Structures in Educational Change: Prigogine and the Academy Donald L. Gilstrap Wichita State University Citation Gilstrap, Donald L. 2007. Dissipative structures in educational change: Prigogine and the academy. -- International Journal of Leadership in Education: Theory and Practice; v.10 no.1, pp. 49-69. A post-print of this paper is posted in the Shocker Open Access Repository: http://soar.wichita.edu/handle/10057/6944 Dissipative structures in educational change: Prigogine and the academy Donald L. Gilstrap This article is an interpretive study of the theory of irreversible and dissipative systems process transformation of Nobel Prize winning physicist Ilya Prigogine and how it relates to the phenomenological study of leadership and organizational change in educational settings. Background analysis on the works of Prigogine is included as a foundation for human inquiry and metaphor generation for open, dissipative systems in educational settings. International case study research by contemporary systems and leadership theorists on dissipative structures theory has also been included to form the interpretive framework for exploring alternative models of leadership theory in far from equilibrium educational settings. Interpretive analysis explores the metaphorical significance, connectedness, and inference of dissipative systems and helps further our knowledge of human-centered transformations in schools and colleges. Introduction Over the course of the past 30 years, educational institutions have undergone dramatic shifts, leading teachers and administrators in new and sometimes competing directions. We are all too familiar with rising enrolments and continual setbacks in financial resources. Perhaps this has never been more evident, particularly in the USA, than with the recent changes in educational and economic policies. -

A Complete Guide to Benzene
A Complete Guide to Benzene Benzene is an important organic chemical compound with the chemical formula C6H6. The benzene molecule is composed of six carbon atoms joined in a ring with one hydrogen atom attached to each. As it contains only carbon and hydrogen atoms, benzene is classed as a hydrocarbon. Benzene is a natural constituent of crude oil and is one of the elementary petrochemicals. Due to the cyclic continuous pi bond between the carbon atoms, benzene is classed as an aromatic hydrocarbon, the second [n]- annulene ([6]-annulene). It is sometimes abbreviated Ph–H. Benzene is a colourless and highly flammable liquid with a sweet smell. Source: Wikipedia Protecting people and the environment Protecting both people and the environment whilst meeting the operational needs of your business is very important and, if you have operations in the UK you will be well aware of the requirements of the CoSHH Regulations1 and likewise the Code of Federal Regulations (CFR) in the US2. Similar legislation exists worldwide, the common theme being an onus on hazard identification, risk assessment and the provision of appropriate control measures (bearing in mind the hierarchy of controls) as well as health surveillance in most cases. And whilst toxic gasses such as hydrogen sulphide and carbon monoxide are a major concern because they pose an immediate (acute) danger to life, long term exposure to relatively low level concentrations of other gasses or vapours such as volatile organic compounds (VOC) are of equal importance because of the chronic illnesses that can result from that ongoing exposure. Benzene, a common VOC Organic means the chemistry of carbon based compounds, which are substances that results from a combination of two or more different chemical elements. -

Quantum Reform
feature Quantum reform Leonie Mueck Quantum computers potentially offer a faster way to calculate chemical properties, but the exact implications of this speed-up have only become clear over the last year. The first quantum computers are likely to enable calculations that cannot be performed classically, which might reform quantum chemistry — but we should not expect a revolution. t had been an exhausting day at the 2012 International Congress of Quantum IChemistry with countless presentations reporting faster and more accurate methods for calculating chemical information using computers. In the dry July heat of Boulder, Colorado, a bunch of young researchers decided to end the day with a cool beer, but the scientific discussion didn’t fade away. Thoughts on the pros and cons of all the methods — the approximations they involved and the chemical problems that they could solve — bounced across the table, until somebody said “Anyway, in a few years we will have a quantum computer and our approximate methods will be obsolete”. An eerie silence followed. What a frustrating thought! They were devoting their careers to quantum chemistry — working LIBRARY PHOTO RICHARD KAIL/SCIENCE tirelessly on applying the laws of quantum mechanics to treat complex chemical of them offering a different trade-off between cannot do on a ‘classical’ computer, it does problems with a computer. And in one fell accuracy and computational feasibility. not look like all the conventional quantum- swoop, would all of those efforts be wasted Enter the universal quantum computer. chemical methods will become obsolete or as chemists turn to quantum computers and This dream machine would basically work that quantum chemists will be out of their their enormous computing power? like a normal digital computer; it would jobs in the foreseeable future.