Ana Isabel Borges Ferraz
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Broad Poster Vivek
A novel computational method for finding regions with copy number abnormalities in cancer cells Vivek, Manuel Garber, and Mike Zody Broad Institute of MIT and Harvard, Cambridge, MA, USA Introduction Results Cancer can result from the over expression of oncogenes, genes which control and regulate cell growth. Sometimes oncogenes increase in 1 2 3 activity due to a specific genetic mutation called a translocation (Fig 1). SMAD4 – a gene known to be deleted in pancreatic COX10 – a gene deleted in cytochrome c oxidase AK001392 – a hereditary prostate cancer protein This translocation allows the oncogene to remain as active as its paired carcinoma deficiency, known to be related to cell proliferation gene. Amplification of this mutation can occur, thereby creating the proper conditions for uncontrolled cell growth; consequently, each Results from Analysis Program Results from Analysis Program Results from Analysis Program component of the translocation will amplify in similar quantities. In this mutation, the chromosomal region containing the oncogene displaces to Region 1 Region 2 R2 Region 1 Region 2 R2 Region 1 Region 2 R2 a region on another chromosome containing a gene that is expressed Chr18:47044749-47311978 Chr17:13930739-14654741 0.499070821478475 Chr17:13930739-14654741 Chr18:26861790-27072166 0.47355172850856 Chr17:12542326-13930738 Chr8:1789292-1801984 0.406208680312004 frequently. Actual region containing gene Actual region containing gene Actual region containing gene chr18: 45,842,214 - 48,514,513 chr17: 13,966,862 - 14,068,461 chr17: 12,542,326 - 13,930,738 Fig 1. Two chromosomal regions (abcdef and ghijk) are translocating to create two new regions (abckl and ghijedf). -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Aneuploidy: Using Genetic Instability to Preserve a Haploid Genome?
Health Science Campus FINAL APPROVAL OF DISSERTATION Doctor of Philosophy in Biomedical Science (Cancer Biology) Aneuploidy: Using genetic instability to preserve a haploid genome? Submitted by: Ramona Ramdath In partial fulfillment of the requirements for the degree of Doctor of Philosophy in Biomedical Science Examination Committee Signature/Date Major Advisor: David Allison, M.D., Ph.D. Academic James Trempe, Ph.D. Advisory Committee: David Giovanucci, Ph.D. Randall Ruch, Ph.D. Ronald Mellgren, Ph.D. Senior Associate Dean College of Graduate Studies Michael S. Bisesi, Ph.D. Date of Defense: April 10, 2009 Aneuploidy: Using genetic instability to preserve a haploid genome? Ramona Ramdath University of Toledo, Health Science Campus 2009 Dedication I dedicate this dissertation to my grandfather who died of lung cancer two years ago, but who always instilled in us the value and importance of education. And to my mom and sister, both of whom have been pillars of support and stimulating conversations. To my sister, Rehanna, especially- I hope this inspires you to achieve all that you want to in life, academically and otherwise. ii Acknowledgements As we go through these academic journeys, there are so many along the way that make an impact not only on our work, but on our lives as well, and I would like to say a heartfelt thank you to all of those people: My Committee members- Dr. James Trempe, Dr. David Giovanucchi, Dr. Ronald Mellgren and Dr. Randall Ruch for their guidance, suggestions, support and confidence in me. My major advisor- Dr. David Allison, for his constructive criticism and positive reinforcement. -

Effet De La Cryptorchidie Sur Le Transcriptome Testiculaire Humain
MARIE EVE BERGERON EFFET DE LA CRYPTORCHIDIE SUR LE TRANSCRIPTOME TESTICULAIRE HUMAIN Mémoire présenté à la Faculté des études supérieures et postdoctorales de l’Université Laval dans le cadre du programme de maîtrise en Physiologie-Endocrinologie pour l’obtention du grade de Maître ès sciences (M.Sc.) DÉPARTEMENT D’OBSTÉTRIQUE ET DE GYNÉCOLOGIE FACULTÉ DE MÉDECINE UNIVERSITÉ LAVAL QUÉBEC 2012 © Marie Eve Bergeron, 2012 Résumé Les niveaux d’expression de nombreux gènes peuvent être affectés par l’environnement et mener au développement de la cryptorchidie. Cette malformation congénitale est la plus commune dont une des conséquences majeures est l’infertilité masculine due au testicule non-descendu, auquel un risque plus élevé de cancer testiculaire est associé. L’expression des ARN totaux isolés à partir de biopsies testiculaires ont été analysés par micropuces, puis par une analyse bio-informatique et une validation par RT-qPCR de plusieurs gènes sélectionnés. Ces analyses m’ont permis d’identifier plus de deux milles candidats montrant une expression différente entre des sujets cryptorchides et normaux. Certains de ces gènes sélectionnés peuvent être associés à la descente testiculaire, d’autres au cancer testiculaire ou encore aux divers types cellulaires retrouvés dans cet organe. Les différences dans le transcriptome dues à la cryptorchidie vont nous aider à comprendre la cause génétique de cette maladie. ii Abstract Expression level of numerous genes may be affected by environmental condition and lead to development of cryptorchidism. The most common congenital malformation in male is cryptorchidism. One major consequence of this anomaly is infertility due to undescended testis, to which an increased risk of testicular cancer is associated. -

Supplemental Figure S1 Differentially Methylated Regions (Dmrs
Supplemental Figure S1 '$$#0#,2'**7+#2&7*2#"0#%'-,11 #25##,"'1#1#122#1 '!2-0'*"#.'!2'-,-$122,1'2'-,$0-+2- !"Q !"2-$%," $ 31',% 25-$-*" !&,%# ," ' 0RTRW 1 !32V-$$ !0'2#0'T - #.0#1#,22'-, -$ "'$$#0#,2'**7+#2&7*2#"%#,#11',.0#,2#1,"2&#'0 #&4'-022,1'2'-, #25##,"'$$#0#,2"'1#1#122#1T-*!)00-51',"'!2#&7.#0+#2&7*2#"%#,#1Q%0700-51 &7.-+#2&7*2#"%#,#1Q31',%25-$-*"!&,%#,"'0RTRW1!32V-$$!0'2#0'T-%#,#1 +#22&# -4#!0'2#0'22,1'2'-,$0-+$%2-$Q5#2�#$-0#*1-',!*3"#" %#,#15'2&V4*3#0RTRWT$$#!2#"%#,10#&'%&*'%&2#" 712#0'1)1#T Supplemental Figure S2 Validation of results from the HELP assay using Epityper MassarrayT #13*21 $0-+ 2&# 1$ 117 5#0# !-00#*2#" 5'2& /3,2'22'4# +#2&7*2'-, ,*78#" 7 '13*$'2#11007$-04V-,"6U-%#,#.0-+-2#00#%'-,1T11007 51.#0$-0+#"31',%**4'* *#1+.*#1T S Supplemental Fig. S1 A unique hypermethylated genes (methylation sites) 454 (481) 5693 (6747) 120 (122) NLMGUS NEWMM REL 2963 (3207) 1338 (1560) 5 (5) unique hypomethylated genes (methylation sites) B NEWMM 0 (0) MGUS 454 (481) 0 (0) NEWMM REL NL 3* (2) 2472 (3066) NEWMM 2963 REL (3207) 2* (2) MGUS 0 (0) REL 2 (2) NEWMM 0 (0) REL Supplemental Fig. S2 A B ARID4B DNMT3A Methylation by MassArray Methylation by MassArray 0 0.2 0.4 0.6 0.8 1 1.2 0.5 0.6 0.7 0.8 0.9 1 2 0 NL PC MGUS 1.5 -0.5 NEW MM 1 REL MM -1 0.5 -1.5 0 -2 -0.5 -1 -2.5 -1.5 -3 Methylation by HELP Assay Methylation by HELP Methylation by HELP Assay Methylation by HELP -2 -3.5 -2.5 -4 Supplemental tables "3..*#+#,2*6 *#"SS 9*','!*!&0!2#0'12'!1-$.2'#,21+.*#1 DZ_STAGE Age Gender Ethnicity MM isotype PCLI Cytogenetics -

Original Article a Database and Functional Annotation of NF-Κb Target Genes
Int J Clin Exp Med 2016;9(5):7986-7995 www.ijcem.com /ISSN:1940-5901/IJCEM0019172 Original Article A database and functional annotation of NF-κB target genes Yang Yang, Jian Wu, Jinke Wang The State Key Laboratory of Bioelectronics, Southeast University, Nanjing 210096, People’s Republic of China Received November 4, 2015; Accepted February 10, 2016; Epub May 15, 2016; Published May 30, 2016 Abstract: Backgrounds: The previous studies show that the transcription factor NF-κB always be induced by many inducers, and can regulate the expressions of many genes. The aim of the present study is to explore the database and functional annotation of NF-κB target genes. Methods: In this study, we manually collected the most complete listing of all NF-κB target genes identified to date, including the NF-κB microRNA target genes and built the database of NF-κB target genes with the detailed information of each target gene and annotated it by DAVID tools. Results: The NF-κB target genes database was established (http://tfdb.seu.edu.cn/nfkb/). The collected data confirmed that NF-κB maintains multitudinous biological functions and possesses the considerable complexity and diversity in regulation the expression of corresponding target genes set. The data showed that the NF-κB was a central regula- tor of the stress response, immune response and cellular metabolic processes. NF-κB involved in bone disease, immunological disease and cardiovascular disease, various cancers and nervous disease. NF-κB can modulate the expression activity of other transcriptional factors. Inhibition of IKK and IκBα phosphorylation, the decrease of nuclear translocation of p65 and the reduction of intracellular glutathione level determined the up-regulation or down-regulation of expression of NF-κB target genes. -

Supplementary Table 1. Mutated Genes That Contain Protein Domains Identified Through Mutation Enrichment Analysis
Supplementary Table 1. Mutated genes that contain protein domains identified through mutation enrichment analysis A. Breast cancers InterPro ID Mutated genes (number of mutations) IPR000219 ARHGEF4(2), ECT2(1), FARP1(1), FLJ20184(1), MCF2L2(1), NET1(1), OBSCN(5), RASGRF2(2), TRAD(1), VAV3(1) IPR000225 APC2(2), JUP(1), KPNA5(2), SPAG6(1) IPR000357 ARFGEF2(2), CMYA4(1), DRIM(2), JUP(1), KPNA5(2), PIK3R4(1), SPAG6(1) IPR000533 AKAP9(2), C10orf39(1), C20orf23(1), CUTL1(1), HOOK1(1), HOOK3(1), KTN1(2), LRRFIP1(3), MYH1(3), MYH9(2), NEF3(1), NF2(1), RSN(1), TAX1BP1(1), TPM4(1) IPR000694 ADAM12(3), ADAMTS19(1), APC2(2), APXL(1), ARID1B(1), BAT2(2), BAT3(1), BCAR1(1), BCL11A(2), BCORL1(1), C14orf155(3), C1orf2(1), C1QB(1), C6orf31(1), C7orf11(1), CD2(1), CENTD3(3), CHD5(3), CIC(3), CMYA1(2), COL11A1(3), COL19A1(2), COL7A1(3), DAZAP1(1), DBN1(3), DVL3(1), EIF5(1), FAM44A(1), FAM47B(1), FHOD1(1), FLJ20584(1), G3BP2(2), GAB1(2), GGA3(1), GLI1(3), GPNMB(2), GRIN2D(3), HCN3(1), HOXA3(2), HOXA4(1), IRS4(1), KCNA5(1), KCNC2(1), LIP8(1), LOC374955(1), MAGEE1(2), MICAL1(2), MICAL‐L1(1), MLLT2(1), MMP15(1), N4BP2(1), NCOA6(2), NHS(1), NUP214(3), ODZ1(3), PER1(2), PER2(1), PHC1(1), PLXNB1(1), PPM1E(2), RAI17(2), RAPH1(2), RBAF600(2), SCARF2(1), SEMA4G(1), SLC16A2(1), SORBS1(1), SPEN(2), SPG4(1), TBX1(1), TCF1(2), TCF7L1(1), TESK1(1), THG‐1(1), TP53(18), TRIF(1), ZBTB3(2), ZNF318(2) IPR000909 CENTB1(2), PLCB1(1), PLCG1(1) IPR000998 AEGP(3), EGFL6(2), PRSS7(1) IPR001140 ABCB10(2), ABCB6(1), ABCB8(2) IPR001164 ARFGAP3(1), CENTB1(2), CENTD3(3), CENTG1(2) IPR001589 -
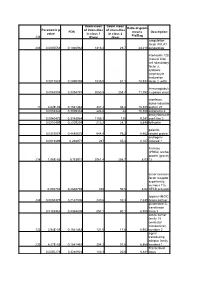
238 Parametric P- Value FDR Geom Mean of Intensities in Class 1 (Plate
Geom mean Geom mean Ratio of geom Parametric p- of intensities of intensities FDR means Description value in class 1 in class 2 Pla/Bag 238 (Plate) (Bag) coagulation factor XIII, A1 206 0,0005558 0,1940962 1313,3 29,7 44,219 polypeptide interleukin 12B (natural killer cell stimulatory factor 2, cytotoxic lymphocyte maturation 0,0017883 0,2490398 1535,5 81,1 18,933 factor 2, p40) Immunoglobuli 0,0063804 0,3394787 3050,5 258,7 11,792 n epsilon chain interferon, alpha-inducible 11 4,62E-05 0,1541482 347,2 33,4 10,395 protein 27 0,0035346 0,3039332 226,8 22 10,309 astrotactin 2 deoxyribonucle 0,0040473 0,3148984 1155,1 125 9,241 ase I-like 3 0,0010495 0,2205294 213,2 24,1 8,846 follistatin galectin- 0,0315078 0,4885675 644,8 76,2 8,462 related protein androgen- 0,0013899 0,238971 287 35,4 8,107 induced 1 A kinase (PRKA) anchor protein (gravin) 236 1,06E-05 0,115911 2061,4 256,7 8,03 12 tumor necrosis factor receptor superfamily, member 11a, 0,002746 0,2846759 405 50,5 8,02 NFKB activator lipoma HMGIC 248 0,0008375 0,2147006 240,6 32,3 7,449 fusion partner glutathione S- transferase 0,0183462 0,4368809 557,1 80,1 6,955 theta 1 solute carrier family 18 (vesicular monoamine), 122 3,94E-05 0,1541482 121,5 17,6 6,903 member 2 signal transducing adaptor family 233 6,27E-05 0,1541482 354,3 51,6 6,866 member 1 Transcribed 0,0050276 0,3240923 140,3 20,6 6,811 locus aldehyde dehydrogenas e 5 family, member A1 (succinate- semialdehyde dehydrogenas 0,0294865 0,4819829 236,6 38,2 6,194 e) guanylate cyclase activator 1A 0,0074099 0,3504479 339,1 55 6,165 -

1 Novel Expression Signatures Identified by Transcriptional Analysis
ARD Online First, published on October 7, 2009 as 10.1136/ard.2009.108043 Ann Rheum Dis: first published as 10.1136/ard.2009.108043 on 7 October 2009. Downloaded from Novel expression signatures identified by transcriptional analysis of separated leukocyte subsets in SLE and vasculitis 1Paul A Lyons, 1Eoin F McKinney, 1Tim F Rayner, 1Alexander Hatton, 1Hayley B Woffendin, 1Maria Koukoulaki, 2Thomas C Freeman, 1David RW Jayne, 1Afzal N Chaudhry, and 1Kenneth GC Smith. 1Cambridge Institute for Medical Research and Department of Medicine, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK 2Roslin Institute, University of Edinburgh, Roslin, Midlothian, EH25 9PS, UK Correspondence should be addressed to Dr Paul Lyons or Prof Kenneth Smith, Department of Medicine, Cambridge Institute for Medical Research, Addenbrooke’s Hospital, Hills Road, Cambridge, CB2 0XY, UK. Telephone: +44 1223 762642, Fax: +44 1223 762640, E-mail: [email protected] or [email protected] Key words: Gene expression, autoimmune disease, SLE, vasculitis Word count: 2,906 The Corresponding Author has the right to grant on behalf of all authors and does grant on behalf of all authors, an exclusive licence (or non-exclusive for government employees) on a worldwide basis to the BMJ Publishing Group Ltd and its Licensees to permit this article (if accepted) to be published in Annals of the Rheumatic Diseases and any other BMJPGL products to exploit all subsidiary rights, as set out in their licence (http://ard.bmj.com/ifora/licence.pdf). http://ard.bmj.com/ on September 29, 2021 by guest. Protected copyright. 1 Copyright Article author (or their employer) 2009. -

Algorithms and Applications of Next-Generation DNA Sequencing
Algorithms and Applications of Next-Generation DNA Sequencing Chip-Seq, database of human variations, and analysis of mammary ductal carcinomas by Anthony Peter Fejes Bachelor of Science, Biochemistry (Hons. Co-op), University of Waterloo, 2000 Bachelor of Independent Studies, University of Waterloo, 2001 Master of Science, Microbiology & Immunology, The University of British Columbia, 2004 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF Doctor of Philosophy in THE FACULTY OF GRADUATE STUDIES (Bioinformatics) The University Of British Columbia (Vancouver) April 2012 © Anthony Peter Fejes, 2012 Abstract Next Generation Sequencing (NGS) technologies enable Deoxyribonucleic Acid (DNA) or Ribonucleic Acid (RNA) sequencing to be done at volumes and speeds several orders of magnitude faster than Sanger (dideoxy termination) based methods and have enabled the development of novel experiment types that would not have been practical before the advent of the NGS-based machines. The dramatically increased throughput of these new protocols requires significant changes to the algorithms used to process and analyze the results. In this thesis, I present novel algorithms used for Chromatin Immunoprecipitation and Sequencing (ChIP-Seq) as well as the structures required and challenges faced for working with Single Nucleotide Variations (SNVs) across a large collection of samples, and finally, I present the results obtained when performing an NGS based analysis of eight mammary ductal carcinoma cell lines and four matched normal cell lines. ii Preface The work described in this thesis is based entirely upon research done at the Canada’s Michael Smith Genome Sciences Centre (BCGSC) in Dr. Steve J.M. Jones’ group by Anthony Fejes. -

Supplementary Table 1: Genes Located on Chromosome 18P11-18Q23, an Area Significantly Linked to TMPRSS2-ERG Fusion
Supplementary Table 1: Genes located on Chromosome 18p11-18q23, an area significantly linked to TMPRSS2-ERG fusion Symbol Cytoband Description LOC260334 18p11 HSA18p11 beta-tubulin 4Q pseudogene IL9RP4 18p11.3 interleukin 9 receptor pseudogene 4 LOC100132166 18p11.32 hypothetical LOC100132166 similar to Rho-associated protein kinase 1 (Rho- associated, coiled-coil-containing protein kinase 1) (p160 LOC727758 18p11.32 ROCK-1) (p160ROCK) (NY-REN-35 antigen) ubiquitin specific peptidase 14 (tRNA-guanine USP14 18p11.32 transglycosylase) THOC1 18p11.32 THO complex 1 COLEC12 18pter-p11.3 collectin sub-family member 12 CETN1 18p11.32 centrin, EF-hand protein, 1 CLUL1 18p11.32 clusterin-like 1 (retinal) C18orf56 18p11.32 chromosome 18 open reading frame 56 TYMS 18p11.32 thymidylate synthetase ENOSF1 18p11.32 enolase superfamily member 1 YES1 18p11.31-p11.21 v-yes-1 Yamaguchi sarcoma viral oncogene homolog 1 LOC645053 18p11.32 similar to BolA-like protein 2 isoform a similar to 26S proteasome non-ATPase regulatory LOC441806 18p11.32 subunit 8 (26S proteasome regulatory subunit S14) (p31) ADCYAP1 18p11 adenylate cyclase activating polypeptide 1 (pituitary) LOC100130247 18p11.32 similar to cytochrome c oxidase subunit VIc LOC100129774 18p11.32 hypothetical LOC100129774 LOC100128360 18p11.32 hypothetical LOC100128360 METTL4 18p11.32 methyltransferase like 4 LOC100128926 18p11.32 hypothetical LOC100128926 NDC80 homolog, kinetochore complex component (S. NDC80 18p11.32 cerevisiae) LOC100130608 18p11.32 hypothetical LOC100130608 structural maintenance -

Diagnostics of Halitosis Complaints by a Multidisciplinary Team
JOP. J Pancreas (Online) 2014 Sep 28; 15(5): 465-474 ORIGINAL ARTICLE Nicotine Alters the Proteome of Two Human Pancreatic Duct Cell Lines Joao A Paulo Department of Cell Biology, Harvard Medical School, Boston, USA ABSTRACT Context Cigarette smoking is a known risk factor of pancreatic disease. Nicotine - a major cigarette tobacco component - can adenocarcinoma, respectively. However, at the biomolecular level, particularly in pancreatic research, the effects of nicotine remaintraffic through unresolved. the circulatory Methods system The effects and may of nicotine induce fibrosis on the andproteomes metastasis, of two hallmarks pancreatic of chronic duct cellpancreatitis lines–an and immortalized pancreatic normal cell line (HPNE) and a cancer cell line (PanC1)- were investigated using mass spectrometry-based proteomics. For each cell line, the global proteomesof cells exposed to nicotine for 24 hrswere compared with untreated cells in triplicate using 6-plex tandem mass tag-based isobaric labeling techniques. Results Over 5,000 proteins were detectedper cell line. Of treatment, 57 of which were so in both cell lines. Amyloid precursor protein, previously observed to increase expression in pancreaticthese, over stellate 900 proteins cells when were exposed differentially to nicotine, abundant was alsowith up-regulated statistical significance in both cell (corrected lines.In general, p-value the <0.01) two cell upon lines nicotine varied in the classes of proteins altered by nicotine treatment, supporting published evidence that nicotine may play different roles in the initiation and progression of pancreatic disease. Conclusions Understanding the underlying mechanisms associating nicotine with pancreatic function is paramount to intervention aiming to retard, arrest, or ameliorate pancreatic disease.