Binnenwerk Cindy Postma.Indd
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Multi‑Resolution Functional Summarization and Alignment of Biological Network Models
This document is downloaded from DR‑NTU (https://dr.ntu.edu.sg) Nanyang Technological University, Singapore. Multi‑resolution functional summarization and alignment of biological network models Seah, Boon Siew 2014 Seah, B. S. (2014). Multi‑resolution functional summarization and alignment of biological network models. Doctoral thesis, Nanyang Technological University, Singapore. https://hdl.handle.net/10356/60641 https://doi.org/10.32657/10356/60641 Downloaded on 07 Oct 2021 12:44:34 SGT ATTENTION: The Singapore Copyright Act applies to the use of this document. Nanyang Technological University Library Multi-resolution Functional Summarization and Alignment of Biological Network Models Seah Boon Siew A THESIS SUBMITTED FOR THE DEGREE OF DOCTOR OF PHILOSOPHY IN COMPUTATION AND SYSTEMS BIOLOGY (CSB) SINGAPORE-MIT ALLIANCE NANYANG TECHNOLOGICAL UNIVERSITY Feb 17, 2014 ATTENTION: The Singapore Copyright Act applies to the use of this document. Nanyang Technological University Library ATTENTION: The Singapore Copyright Act applies to the use of this document. Nanyang Technological University Library DECLARATION I hereby declare that this thesis is my original work and it has been written by me in its entirety. I have duly acknowledged all the sources of information which have been used in the thesis. This thesis has also not been submitted for any degree in any university previously. Seah Boon Siew Feb 17, 2014 ii ATTENTION: The Singapore Copyright Act applies to the use of this document. Nanyang Technological University Library Acknowledgments I would like to thank my supervisor Assoc. Prof. Sourav S. Bhowmick (NTU) and my co-supervisor Prof. C. Forbes Dewey, Jr. (MIT) for their guidance and support. -

(12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer Et Al
USOO8440393B2 (12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer et al. (45) Date of Patent: May 14, 2013 (54) PRO-ANGIOGENIC GENES IN OVARIAN OTHER PUBLICATIONS TUMORENDOTHELIAL CELL, SOLATES Boyd (The Basic Science of Oncology, 1992, McGraw-Hill, Inc., p. (75) Inventors: Michael J. Birrer, Mt. Airy, MD (US); 379). Tomas A. Bonome, Washington, DC Tockman et al. (Cancer Res., 1992, 52:2711s-2718s).* (US); Anil Sood, Pearland, TX (US); Pritzker (Clinical Chemistry, 2002, 48: 1147-1150).* Chunhua Lu, Missouri City, TX (US) Benedict et al. (J. Exp. Medicine, 2001, 193(1) 89-99).* Jiang et al. (J. Biol. Chem., 2003, 278(7) 4763-4769).* (73) Assignees: The United States of America as Matsushita et al. (FEBS Letters, 1999, vol. 443, pp. 348-352).* Represented by the Secretary of the Singh et al. (Glycobiology, 2001, vol. 11, pp. 587-592).* Department of Health and Human Abbosh et al. (Cancer Res. Jun. 1, 2006 66:5582-55.91 and Supple Services, Washington, DC (US); The mental Figs. S1-S7).* University of MD Anderson Cancer Zhai et al. (Chinese General Practice Aug. 2008, 11(8A): 1366 Center, Houston, TX (US) 1367).* Lu et al. (Cancer Res. Feb. 15, 2007, 64(4): 1757-1768).* (*) Notice: Subject to any disclaimer, the term of this Bagnato et al., “Activation of Mitogenic Signaling by Endothelin 1 in patent is extended or adjusted under 35 Ovarian Carcinoma Cells', Cancer Research, vol. 57, pp. 1306-1311, U.S.C. 154(b) by 194 days. 1997. Bouras et al., “Stanniocalcin 2 is an Estrogen-responsive Gene (21) Appl. -

Supplemental Table 3 Site ID Intron Poly(A) Site Type NM/KG Inum
Supplemental Table 3 Site ID Intron Poly(A) site Type NM/KG Inum Region Gene ID Gene Symbol Gene Annotation Hs.120277.1.10 chr3:170997234:170996860 170996950 b NM_153353 7 CDS 151827 LRRC34 leucine rich repeat containing 34 Hs.134470.1.27 chr17:53059664:53084458 53065543 b NM_138962 10 CDS 124540 MSI2 musashi homolog 2 (Drosophila) Hs.162889.1.18 chr14:80367239:80329208 80366262 b NM_152446 12 CDS 145508 C14orf145 chromosome 14 open reading frame 145 Hs.187898.1.27 chr22:28403623:28415294 28404458 b NM_181832 16 3UTR 4771 NF2 neurofibromin 2 (bilateral acoustic neuroma) Hs.228320.1.6 chr10:115527009:115530350 115527470 b BC036365 5 CDS 79949 C10orf81 chromosome 10 open reading frame 81 Hs.266308.1.2 chr11:117279579:117278191 117278967 b NM_032046 12 CDS 84000 TMPRSS13 transmembrane protease, serine 13 Hs.266308.1.4 chr11:117284536:117281662 117283722 b NM_032046 9 CDS 84000 TMPRSS13 transmembrane protease, serine 13 Hs.2689.1.4 chr10:53492398:53563605 53492622 b NM_006258 7 CDS 5592 PRKG1 protein kinase, cGMP-dependent, type I Hs.280781.1.6 chr18:64715646:64829150 64715837 b NM_024781 4 CDS 79839 C18orf14 chromosome 18 open reading frame 14 Hs.305985.2.25 chr12:8983686:8984438 8983942 b BX640639 17 3UTR NA NA NA Hs.312098.1.36 chr1:151843991:151844258 151844232 b NM_003815 15 CDS 8751 ADAM15 a disintegrin and metalloproteinase domain 15 (metargidin) Hs.314338.1.11 chr21:39490293:39481214 39487623 b NM_018963 41 CDS 54014 BRWD1 bromodomain and WD repeat domain containing 1 Hs.33368.1.3 chr15:92685158:92689361 92688314 b NM_018349 6 CDS 55784 MCTP2 multiple C2-domains with two transmembrane regions 2 Hs.346736.1.21 chr2:99270738:99281614 99272414 b AK126402 10 3UTR 51263 MRPL30 mitochondrial ribosomal protein L30 Hs.445061.1.19 chr16:69322898:69290216 69322712 b NM_018052 14 CDS 55697 VAC14 Vac14 homolog (S. -

Rattlesnake Genome Supplemental Materials 1 SUPPLEMENTAL
Rattlesnake Genome Supplemental Materials 1 1 SUPPLEMENTAL MATERIALS 2 Table of Contents 3 1. Supplementary Methods …… 2 4 2. Supplemental Tables ……….. 23 5 3. Supplemental Figures ………. 37 Rattlesnake Genome Supplemental Materials 2 6 1. SUPPLEMENTARY METHODS 7 Prairie Rattlesnake Genome Sequencing and Assembly 8 A male Prairie Rattlesnake (Crotalus viridis viridis) collected from a wild population in Colorado was 9 used to generate the genome sequence. This specimen was collected and humanely euthanized according 10 to University of Northern Colorado Institutional Animal Care and Use Committee protocols 0901C-SM- 11 MLChick-12 and 1302D-SM-S-16. Colorado Parks and Wildlife scientific collecting license 12HP974 12 issued to S.P. Mackessy authorized collection of the animal. Genomic DNA was extracted using a 13 standard Phenol-Chloroform-Isoamyl alcohol extraction from liver tissue that was snap frozen in liquid 14 nitrogen. Multiple short-read sequencing libraries were prepared and sequenced on various platforms, 15 including 50bp single-end and 150bp paired-end reads on an Illumina GAII, 100bp paired-end reads on an 16 Illumina HiSeq, and 300bp paired-end reads on an Illumina MiSeq. Long insert libraries were also 17 constructed by and sequenced on the PacBio platform. Finally, we constructed two sets of mate-pair 18 libraries using an Illumina Nextera Mate Pair kit, with insert sizes of 3-5 kb and 6-8 kb, respectively. 19 These were sequenced on two Illumina HiSeq lanes with 150bp paired-end sequencing reads. Short and 20 long read data were used to assemble the previous genome assembly version CroVir2.0 (NCBI accession 21 SAMN07738522). -

The DNA Sequence and Comparative Analysis of Human Chromosome 20
articles The DNA sequence and comparative analysis of human chromosome 20 P. Deloukas, L. H. Matthews, J. Ashurst, J. Burton, J. G. R. Gilbert, M. Jones, G. Stavrides, J. P. Almeida, A. K. Babbage, C. L. Bagguley, J. Bailey, K. F. Barlow, K. N. Bates, L. M. Beard, D. M. Beare, O. P. Beasley, C. P. Bird, S. E. Blakey, A. M. Bridgeman, A. J. Brown, D. Buck, W. Burrill, A. P. Butler, C. Carder, N. P. Carter, J. C. Chapman, M. Clamp, G. Clark, L. N. Clark, S. Y. Clark, C. M. Clee, S. Clegg, V. E. Cobley, R. E. Collier, R. Connor, N. R. Corby, A. Coulson, G. J. Coville, R. Deadman, P. Dhami, M. Dunn, A. G. Ellington, J. A. Frankland, A. Fraser, L. French, P. Garner, D. V. Grafham, C. Grif®ths, M. N. D. Grif®ths, R. Gwilliam, R. E. Hall, S. Hammond, J. L. Harley, P. D. Heath, S. Ho, J. L. Holden, P. J. Howden, E. Huckle, A. R. Hunt, S. E. Hunt, K. Jekosch, C. M. Johnson, D. Johnson, M. P. Kay, A. M. Kimberley, A. King, A. Knights, G. K. Laird, S. Lawlor, M. H. Lehvaslaiho, M. Leversha, C. Lloyd, D. M. Lloyd, J. D. Lovell, V. L. Marsh, S. L. Martin, L. J. McConnachie, K. McLay, A. A. McMurray, S. Milne, D. Mistry, M. J. F. Moore, J. C. Mullikin, T. Nickerson, K. Oliver, A. Parker, R. Patel, T. A. V. Pearce, A. I. Peck, B. J. C. T. Phillimore, S. R. Prathalingam, R. W. Plumb, H. Ramsay, C. M. -
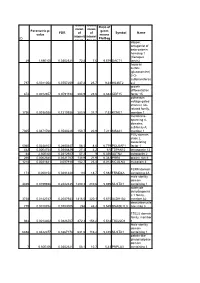
ID Parametric P
Geom Geom Ratio of mean mean Parametric p- geom FDR of of Symbol Name value means intensit intensi ID Pla/Bag ies in ties in dapper, antagonist of beta-catenin, homolog 1 (Xenopus 29 1,69E-05 0,0302321 72,8 7,6 9,579 DACT1 laevis) heparan sulfate (glucosamine) 3-O- sulfotransferas 797 0,0011054 0,0757259 237,2 25,7 9,23 HS3ST2 e 2 growth differentiation 874 0,0012657 0,0791194 204,9 23,6 8,682 GDF15 factor 15 potassium voltage-gated channel, Isk- related family, 1756 0,0038928 0,1210936 283,5 37,7 7,52 KCNE1 member 1 membrane- spanning 4- domains, subfamily A, 7305 0,0471766 0,3530226 150,7 20,9 7,211 MS4A1 member 1 POU domain, class 2, associating 5060 0,0248103 0,2680447 58,3 8,6 6,779 POU2AF1 factor 1 442 0,0004744 0,0586829 27,3 4,2 6,5 TSPAN12 tetraspanin 12 44 2,60E-05 0,0312927 57,2 9 6,356 ASTN2 astrotactin 2 266 0,0002545 0,0521767 138,9 21,9 6,342 PRR6 proline rich 6 1218 0,0021841 0,097918 152,7 25,4 6,012 MCOLN3 mucolipin 3 FERM domain 173 0,000154 0,0481428 110 18,7 5,882 FRMD4A containing 4A male sterility domain 4469 0,0199658 0,2442435 1200,3 210,6 5,699 MLSTD1 containing 1 aldehyde dehydrogenas e 1 family, 3738 0,0142747 0,2087933 1816,5 320,1 5,675 ALDH1A2 member A2 deoxyribonucle 779 0,0010754 0,0753005 268 48,3 5,549 DNASE1L3 ase I-like 3 TSC22 domain family, member 944 0,0014302 0,0826707 872,3 158,2 5,514 TSC22D1 1 Male sterility domain 6858 0,0422377 0,3367178 631,6 116,2 5,435 MLSTD1 containing 1 patatin-like phospholipase domain 7 6,60E-06 0,0302321 58,1 10,7 5,43 PNPLA3 containing 3 hypothetical protein 1857 -

(12) Patent Application Publication (10) Pub. No.: US 2013/0274315 A1 Birrer Et Al
US 201302743 15A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0274315 A1 Birrer et al. (43) Pub. Date: Oct. 17, 2013 (54) PRO-ANGIOGENC GENES IN OVARAN (60) Provisional application No. 60/901,455, filed on Feb. TUMORENDOTHELIAL CELL, SOLATES 14, 2007. (71) Applicants: The University of Texas MD Anderson Publication Classification Cancer Center, Houston, TX (US); The Government of the U.S.A as (51) Int. Cl. represented by the Secretary of the CI2O I/68 (2006.01) Department of He, Rockville, MD (US) (52) U.S. Cl. CPC .................................... CI2O I/6886 (2013.01) (72) Inventors: Michael J. Birrer, Mt. Airy, MD (US); USPC ............... 514/44A: 435/6.12: 506/9: 435/7.1 Tomas A. Bonome, Washington, DC (US); Anil Sood, Pearland, TX (US); (57) ABSTRACT Chunhua Lu, Missouri City, TX (US) A gene profiling signature for ovarian tumor endothelial cells is disclosed herein. The gene signature can be used to diag nosis or prognosis an ovarian tumor, identify agents to treat an (21) Appl. No.: 13/863,219 ovariantumor, to predict the metastatic potential of an ovarian tumor and to determine the effectiveness of ovarian tumor (22) Filed: Apr. 15, 2013 treatments. Thus, methods are provided for identifying agents that can be used to treat ovarian cancer, for determining the effectiveness of an ovarian tumor treatment, or to diagnose or Related U.S. Application Data prognose an ovarian tumor. Methods of treatment are also (60) Division of application No. 12/541,729, filed on Aug. disclosed which include administering a composition that 14, 2009, now Pat. -

Hypoxia As an Evolutionary Force
“The genetic architecture of adaptations to high altitude in Ethiopia” Gorka Alkorta-Aranburu1, Cynthia M. Beall2*, David B. Witonsky1, Amha Gebremedhin3, Jonathan K. Pritchard1,4, Anna Di Rienzo1* 1 Department of Human Genetics, University of Chicago, Chicago, Illinois, United States of America, 2 Department of Anthropology, Case Western Research University, Cleveland, Ohio, United States of America, 3 Department of Internal Medicine, Faculty of Medicine, Addis Ababa University, Addis Ababa, Ethiopia, 4 Howard Hughes Medical Institute * E-mail: [email protected] and [email protected] Corresponding authors: Anna Di Rienzo Department of Human Genetics University of Chicago 920 E. 58th Street Chicago, IL 60637, USA. Cynthia M. Beall Anthropology Department Case Western Reserve University 238 Mather Memorial Building 11220 Bellflower Road Cleveland, OH 44106, USA. 1 ABSTRACT Although hypoxia is a major stress on physiological processes, several human populations have survived for millennia at high altitudes, suggesting that they have adapted to hypoxic conditions. This hypothesis was recently corroborated by studies of Tibetan highlanders, which showed that polymorphisms in candidate genes show signatures of natural selection as well as well-replicated association signals for variation in hemoglobin levels. We extended genomic analysis to two Ethiopian ethnic groups: Amhara and Oromo. For each ethnic group, we sampled low and high altitude residents, thus allowing genetic and phenotypic comparisons across altitudes and across ethnic groups. Genome- wide SNP genotype data were collected in these samples by using Illumina arrays. We find that variants associated with hemoglobin variation among Tibetans or other variants at the same loci do not influence the trait in Ethiopians. -

GENE LIST ANTI-CORRELATED Systematic Common Description
GENE LIST ANTI-CORRELATED Systematic Common Description 210348_at 4-Sep Septin 4 206155_at ABCC2 ATP-binding cassette, sub-family C (CFTR/MRP), member 2 221226_s_at ACCN4 Amiloride-sensitive cation channel 4, pituitary 207427_at ACR Acrosin 214957_at ACTL8 Actin-like 8 207422_at ADAM20 A disintegrin and metalloproteinase domain 20 216998_s_at ADAM5 synonym: tMDCII; Homo sapiens a disintegrin and metalloproteinase domain 5 (ADAM5) on chromosome 8. 216743_at ADCY6 Adenylate cyclase 6 206807_s_at ADD2 Adducin 2 (beta) 208544_at ADRA2B Adrenergic, alpha-2B-, receptor 38447_at ADRBK1 Adrenergic, beta, receptor kinase 1 219977_at AIPL1 211560_s_at ALAS2 Aminolevulinate, delta-, synthase 2 (sideroblastic/hypochromic anemia) 211004_s_at ALDH3B1 Aldehyde dehydrogenase 3 family, member B1 204705_x_at ALDOB Aldolase B, fructose-bisphosphate 220365_at ALLC Allantoicase 204664_at ALPP Alkaline phosphatase, placental (Regan isozyme) 216377_x_at ALPPL2 Alkaline phosphatase, placental-like 2 221114_at AMBN Ameloblastin, enamel matrix protein 206892_at AMHR2 Anti-Mullerian hormone receptor, type II 217293_at ANGPT1 Angiopoietin 1 210952_at AP4S1 Adaptor-related protein complex 4, sigma 1 subunit 207158_at APOBEC1 Apolipoprotein B mRNA editing enzyme, catalytic polypeptide 1 213611_at AQP5 Aquaporin 5 216219_at AQP6 Aquaporin 6, kidney specific 206784_at AQP8 Aquaporin 8 214490_at ARSF Arylsulfatase F 216204_at ARVCF Armadillo repeat gene deletes in velocardiofacial syndrome 214070_s_at ATP10B ATPase, Class V, type 10B 221240_s_at B3GNT4 UDP-GlcNAc:betaGal -
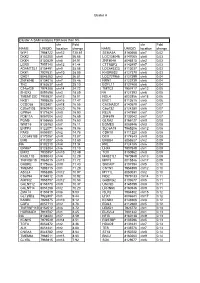
Copy of Supplementary Table 2
Cluster A Cluster A SAM analysis FDR less than 5% chr Fold chr Fold NAME UNIQID location change NAME UNIQID location change WIF1 7964722 chr12 130.61 SEMA3A 8140668 chr7 0.02 DKK2 8102200 chr4 56.66 LOC138046 8147065 chr8 0.03 DKK4 8150529 chr8 54.91 ZNF804A 8046815 chr2 0.03 LGR5 7957140 chr12 41.44 CTTNBP2 8142497 chr7 0.03 ADAMTSL1 8154491 chr9 35.08 LOC645323 8113037 chr5 0.03 DKK1 7927631 chr10 28.09 KHDRBS2 8127370 chr6 0.03 GAD1 8046283 chr2 26.81 LOC727966 8127399 chr6 0.04 ZNF804B 8134018 chr7 25.46 NRN1 8123739 chr6 0.04 TNC 8163637 chr9 25.10 EGFL11 8127408 chr6 0.04 C14orf29 7974288 chr14 24.72 TMTC2 7957417 chr12 0.05 SHOX2 8091698 chr3 18.89 NA 8127393 chr6 0.05 TMEM132C 7959827 chr12 18.01 NOL4 8022856 chr18 0.06 NKD1 7995525 chr16 17.47 ENC1 8112615 chr5 0.06 CCDC68 8023401 chr18 16.55 CACNA2D1 8140579 chr7 0.07 C20orf103 8060940 chr20 16.06 C6orf32 8124280 chr6 0.07 GPR64 8171624 chrX 15.90 RELN 8141950 chr7 0.07 PDE11A 8057004 chr2 15.69 ZNF679 8133042 chr7 0.07 PGM5 8155665 chr9 15.60 GLRA2 8166127 chrX 0.08 WNT16 8135763 chr7 15.11 EOMES 8085946 chr3 0.08 ENPP3 8122071 chr6 15.06 SLC6A15 7965206 chr12 0.08 PAX3 8059301 chr2 14.78 CDH18 8111220 chr5 0.08 LOC645188 8170257 chrX 13.87 DCX 8174543 chrX 0.08 EMB 8112007 chr5 13.60 ERBB4 8058627 chr2 0.09 NA 8102210 chr4 13.34 PRL 8124185 chr6 0.09 EPHA7 8128284 chr6 13.10 LHX4 7907849 chr1 0.09 EMX2 7930857 chr10 12.49 TOX 8150962 chr8 0.09 RASL11B 8095043 chr4 12.29 MAB21L1 7970949 chr13 0.09 TNFRSF19 7968015 chr13 11.72 MPP3 8015846 chr17 0.09 GABRE 8175666 chrX 11.40 -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

Supplemental Table 3 Two-Class Paired Significance Analysis of Microarrays Comparing Gene Expression Between Paired
Supplemental Table 3 Two‐class paired Significance Analysis of Microarrays comparing gene expression between paired pre‐ and post‐transplant kidneys biopsies (N=8). Entrez Fold q‐value Probe Set ID Gene Symbol Unigene Name Score Gene ID Difference (%) Probe sets higher expressed in post‐transplant biopsies in paired analysis (N=1871) 218870_at 55843 ARHGAP15 Rho GTPase activating protein 15 7,01 3,99 0,00 205304_s_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 6,30 4,50 0,00 1563649_at ‐‐ ‐‐ ‐‐ 6,24 3,51 0,00 1567913_at 541466 CT45‐1 cancer/testis antigen CT45‐1 5,90 4,21 0,00 203932_at 3109 HLA‐DMB major histocompatibility complex, class II, DM beta 5,83 3,20 0,00 204606_at 6366 CCL21 chemokine (C‐C motif) ligand 21 5,82 10,42 0,00 205898_at 1524 CX3CR1 chemokine (C‐X3‐C motif) receptor 1 5,74 8,50 0,00 205303_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 5,68 6,87 0,00 226841_at 219972 MPEG1 macrophage expressed gene 1 5,59 3,76 0,00 203923_s_at 1536 CYBB cytochrome b‐245, beta polypeptide (chronic granulomatous disease) 5,58 4,70 0,00 210135_s_at 6474 SHOX2 short stature homeobox 2 5,53 5,58 0,00 1562642_at ‐‐ ‐‐ ‐‐ 5,42 5,03 0,00 242605_at 1634 DCN decorin 5,23 3,92 0,00 228750_at ‐‐ ‐‐ ‐‐ 5,21 7,22 0,00 collagen, type III, alpha 1 (Ehlers‐Danlos syndrome type IV, autosomal 201852_x_at 1281 COL3A1 dominant) 5,10 8,46 0,00 3493///3 IGHA1///IGHA immunoglobulin heavy constant alpha 1///immunoglobulin heavy 217022_s_at 494 2 constant alpha 2 (A2m marker) 5,07 9,53 0,00 1 202311_s_at