Multi‑Resolution Functional Summarization and Alignment of Biological Network Models
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Binnenwerk Cindy Postma.Indd
CHAPTER 6 Multiple putative oncogenes at the chromosome 20q amplicon contribute to colorectal adenoma to carcinoma progression Gut 2009, 58: 79-89 Beatriz Carvalho Cindy Postma Sandra Mongera Erik Hopmans Sharon Diskin Mark A. van de Wiel Wim van Criekinge Olivier Thas Anja Matthäi Miguel A. Cuesta Jochim S. Terhaar sive Droste Mike Craanen Evelin Schröck Bauke Ylstra Gerrit A. Meijer 104 | Chapter 6 Abstract Objective: This study aimed to identify the oncogenes at 20q involved in colorectal adenoma to carcinoma progression by measuring the effect of 20q gain on mRNA expression of genes in this amplicon. Methods: Segmentation of DNA copy number changes on 20q was performed by array CGH in 34 non-progressed colorectal adenomas, 41 progressed adenomas (i.e. adenomas that present a focus of cancer) and 33 adenocarcinomas. Moreover, a robust analysis of altered expression of genes in these segments was performed by microarray analysis in 37 adenomas and 31 adenocarcinomas. Protein expression was evaluated by immunohistochemistry on tissue microarrays. Results: The genes C20orf24, AURKA, RNPC1, TH1L, ADRM1, C20orf20 and TCFL5, mapping at 20q were signifi cantly overexpressed in carcinomas compared to adenomas as consequence of copy number gain of 20q. Conclusion: This approach revealed C20orf24, AURKA, RNPC1, TH1L, ADRM1, C20orf20 and TCFL5 genes to be important in chromosomal instability-related adenoma to carcinoma progression. These genes therefore may serve as highly specifi c biomarkers for colorectal cancer with potential clinical applications. Putative oncogenes at chromosome 20q in colorectal carcinogenesis | 105 Introduction The majority of cancers are epithelial in origin and arise through a stepwise progression from normal cells, through dysplasia, into malignant cells that invade surrounding tissues and have metastatic potential. -

The DNA Sequence and Comparative Analysis of Human Chromosome 20
articles The DNA sequence and comparative analysis of human chromosome 20 P. Deloukas, L. H. Matthews, J. Ashurst, J. Burton, J. G. R. Gilbert, M. Jones, G. Stavrides, J. P. Almeida, A. K. Babbage, C. L. Bagguley, J. Bailey, K. F. Barlow, K. N. Bates, L. M. Beard, D. M. Beare, O. P. Beasley, C. P. Bird, S. E. Blakey, A. M. Bridgeman, A. J. Brown, D. Buck, W. Burrill, A. P. Butler, C. Carder, N. P. Carter, J. C. Chapman, M. Clamp, G. Clark, L. N. Clark, S. Y. Clark, C. M. Clee, S. Clegg, V. E. Cobley, R. E. Collier, R. Connor, N. R. Corby, A. Coulson, G. J. Coville, R. Deadman, P. Dhami, M. Dunn, A. G. Ellington, J. A. Frankland, A. Fraser, L. French, P. Garner, D. V. Grafham, C. Grif®ths, M. N. D. Grif®ths, R. Gwilliam, R. E. Hall, S. Hammond, J. L. Harley, P. D. Heath, S. Ho, J. L. Holden, P. J. Howden, E. Huckle, A. R. Hunt, S. E. Hunt, K. Jekosch, C. M. Johnson, D. Johnson, M. P. Kay, A. M. Kimberley, A. King, A. Knights, G. K. Laird, S. Lawlor, M. H. Lehvaslaiho, M. Leversha, C. Lloyd, D. M. Lloyd, J. D. Lovell, V. L. Marsh, S. L. Martin, L. J. McConnachie, K. McLay, A. A. McMurray, S. Milne, D. Mistry, M. J. F. Moore, J. C. Mullikin, T. Nickerson, K. Oliver, A. Parker, R. Patel, T. A. V. Pearce, A. I. Peck, B. J. C. T. Phillimore, S. R. Prathalingam, R. W. Plumb, H. Ramsay, C. M. -

Hypoxia As an Evolutionary Force
“The genetic architecture of adaptations to high altitude in Ethiopia” Gorka Alkorta-Aranburu1, Cynthia M. Beall2*, David B. Witonsky1, Amha Gebremedhin3, Jonathan K. Pritchard1,4, Anna Di Rienzo1* 1 Department of Human Genetics, University of Chicago, Chicago, Illinois, United States of America, 2 Department of Anthropology, Case Western Research University, Cleveland, Ohio, United States of America, 3 Department of Internal Medicine, Faculty of Medicine, Addis Ababa University, Addis Ababa, Ethiopia, 4 Howard Hughes Medical Institute * E-mail: [email protected] and [email protected] Corresponding authors: Anna Di Rienzo Department of Human Genetics University of Chicago 920 E. 58th Street Chicago, IL 60637, USA. Cynthia M. Beall Anthropology Department Case Western Reserve University 238 Mather Memorial Building 11220 Bellflower Road Cleveland, OH 44106, USA. 1 ABSTRACT Although hypoxia is a major stress on physiological processes, several human populations have survived for millennia at high altitudes, suggesting that they have adapted to hypoxic conditions. This hypothesis was recently corroborated by studies of Tibetan highlanders, which showed that polymorphisms in candidate genes show signatures of natural selection as well as well-replicated association signals for variation in hemoglobin levels. We extended genomic analysis to two Ethiopian ethnic groups: Amhara and Oromo. For each ethnic group, we sampled low and high altitude residents, thus allowing genetic and phenotypic comparisons across altitudes and across ethnic groups. Genome- wide SNP genotype data were collected in these samples by using Illumina arrays. We find that variants associated with hemoglobin variation among Tibetans or other variants at the same loci do not influence the trait in Ethiopians. -

Microarray Screening of Differentially Expressed Genes After Up-Regulating Mir-205 Or
bioRxiv preprint doi: https://doi.org/10.1101/618041; this version posted April 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Microarray screening of differentially expressed genes after up-regulating miR-205 or 2 down-regulating miR-141 in cervical cancer cells 3 Xingyu Fang 1¶ , Tingting Yao 1,2* 4 1Department of Gynecological Oncology, Sun Yat‐sen Memorial Hospital, Sun Yat‐sen 5 University, Guangzhou, China 6 2Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene 7 Regulation, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, China 8 * Corresponding author 9 E-mail address [email protected] (TTY) 10 11 12 13 14 15 16 17 18 19 20 1 bioRxiv preprint doi: https://doi.org/10.1101/618041; this version posted April 26, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 21 Microarray screening of differentially expressed genes after up-regulating miR-205 or 22 down-regulating miR-141 in cervical cancer cells 23 Abstract 24 Cervical cancer is one of the most common gynecological malignancies. However,studies 25 on the expression and molecular mechanism of miR-205 and miR-141 in CC are insufficient 26 recently. -

Note to Users
NOTE TO USERS This reproduction is the best copy available. ® UMI Identifying Mouse Genes Putatively Transcriptionally Regulated by the Glucocorticoid Receptor By Zuojian Tang School of Computer Science McGiII University, Montreal January 2005 A thesis submitted to McGiII University in partial fulfillment of the requirements of the degree of Master of Science ©Zuojian Tang 2005 Library and Bibliothèque et 1+1 Archives Canada Archives Canada Published Heritage Direction du Branch Patrimoine de l'édition 395 Wellington Street 395, rue Wellington Ottawa ON K1A ON4 Ottawa ON K1A ON4 Canada Canada Your file Votre référence ISBN: 0-494-12552-7 Our file Notre référence ISBN: 0-494-12552-7 NOTICE: AVIS: The author has granted a non L'auteur a accordé une licence non exclusive exclusive license allowing Library permettant à la Bibliothèque et Archives and Archives Canada to reproduce, Canada de reproduire, publier, archiver, publish, archive, preserve, conserve, sauvegarder, conserver, transmettre au public communicate to the public by par télécommunication ou par l'Internet, prêter, telecommunication or on the Internet, distribuer et vendre des thèses partout dans loan, distribute and sell th es es le monde, à des fins commerciales ou autres, worldwide, for commercial or non sur support microforme, papier, électronique commercial purposes, in microform, et/ou autres formats. paper, electronic and/or any other formats. The author retains copyright L'auteur conserve la propriété du droit d'auteur ownership and moral rights in et des droits moraux qui protège cette thèse. this thesis. Neither the thesis Ni la thèse ni des extraits substantiels de nor substantial extracts from it celle-ci ne doivent être imprimés ou autrement may be printed or otherwise reproduits sans son autorisation. -

Program and Table of Contents
Program and Table of Contents Helpful Information……………………………………………………………………………………..…5 Industry Support……………………………………………………………………………………………...6 About Us………………………….……………………………………………………………………………...7 Conference Faculty………………………………………………………………………………………………..8 Scripps Continuing Education Annual Course Listing ……………………………………………………...….206 Thursday, March 1, 2012 7:30 a.m. Registration and Breakfast 8:15 a.m. Welcome and Opening Remarks………………………………………………………………….13 Chris Van Gorder, FACHE, President and Chief Executive Officer, Scripps Health Eric J. Topol, MD, Director, Scripps Translational Science Institute 8:30 a.m. How Did Sequencing Our Genomes Change Our Lives?.............................................................15 Joe, Retta, Noah and Alexis Beery Morning Session: Changing Medical Practice – Pharmacogenomics Moderators: Samuel Levy, PhD and Eric Topol, MD 9:00 a.m. Pharmacogenetic testing to optimize treatment of patients with coronary artery disease…….23 Matthew J. Price, MD 9:20 a.m. Exome sequencing to understand pharmacogenomics...................................................................24 Samuel Levy, PhD 9:40 a.m. Hypertension pharmacogenomics – Advances and Challenges…………………………….……25 Julie Johnson, PharmD 10:00 a.m. Break, View Exhibits and Networking 10:20 a.m. Predicting Adverse Events with Commonly Used Drugs………………………………………...26 Michael R. Hayden MB, ChB, PhD 10:40 a.m. Pharmacogenomics to optimize treatment of childhood leukemia……………………………...27 William E. Evans, PharmD 11:00 a.m. Morning Panel Discussion/Q&A Morning Session Speakers and Moderators Hot Topics in Genomic Medicine Moderators: Evan Eichler, PhD and Sarah Murray, PhD 11:25 a.m. Incorporating Genomic Medicine into the Delivery System.....................................……………29 Reed Tuckson, MD 11:50 a.m. DNA Transistors and Handheld Sequencing……………………………………………………..31 Leila Shepherd, PhD 12:15 p.m. Lunch, View Exhibits and Networking 1:00 p.m. Social Networks and Evolution……………………………………………………………………32 James Fowler 1 Program and Table of Contents continued on next page. -

Comprehensive Identification and Annotation of Nonprotein-Coding
Comprehensive Identification and Annotation of Non- protein-coding Transcriptomes from Vertebrates Indicates Most ncRNAs are Regulatory Zhipeng Qu A thesis submitted for the degree of Doctor of Philosophy Discipline of Genetics School of Molecular and Biomedical Science The University of Adelaide October 2012 Table of Contents Contents ........................................................................................................................................I Abstract....................................................................................................................................... II Declaration.................................................................................................................................III Acknowledgements .....................................................................................................................V Chapter 1 Introduction................................................................................................................1 Chapter 2 Bovine ncRNAs Are Abundant, Primarily Intergenic, Conserved and Associated with Regulatory Genes .............................................................................................3 Chapter 3 Identification And Comparative Analysis Of ncRNAs In Human, Mouse And Zebrafish Indicate A Conserved Role In Regulation Of Genes Expressed In Brain ............5 Chapter 4 Re-construction and Annotation of Human Protein-coding and Non-coding RNA Co-expression Networks ....................................................................................................7 -

Synaptic Signal Transduction and Transcriptional Control
Synaptic signal transduction and transcriptional control Thesis by Holly C. Beale In Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy California Institute of Technology Pasadena, California 2010 (Submitted Friday 21st May, 2010) ii c 2010 Holly C. Beale All Rights Reserved iii This thesis is dedicated to Gwen Murdock Pollard, to the many people whose friendship and support exceeded anything I could have anticipated, and to Julia Scheinmann. iv Acknowledgments First I would like to thank my advisor, Mary Kennedy, for the the insights she pro- vided over the past seven years and for the freedom she gave me. The breadth of her scientific interest is inspiring. I would also like to thank the other members of my thesis committee, Paul Patterson, Paul Sternberg and the chair, Henry Lester, for their time and feedback over the years. In particular, I thank Paul Patterson for his writing suggestions, which have transformed how I think about my writing, and Henry Lester for his advice and teaching example. As a body, members of the Kennedy lab have been exemplars of insight, collabora- tion and persistence. Holly Carlisle, Tinh Luong and Tami Ursem were invaluable in the completion of this thesis. Andrew Medina-Marino provided a critical opportunity and example of kindness. Dado Marcora has been terrific geek company, a walking encyclopedia of methods and an asker of fundamental questions. Leslie Schenker has been the silent engine of the lab and is a delight every day. It has been an honor to work with these lab members and others past and present, including Mee Choi, Cheryl Gause, Irene Knuesel, Pat Manzerra, Ward Walkup, and Lori Washburn. -
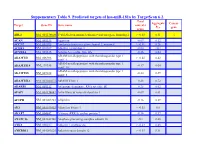
Suppementary Table 9. Predicted Targets of Hsa-Mir-181A by Targetscan 6.2
Suppementary Table 9. Predicted targets of hsa-miR-181a by TargetScan 6.2. Total Aggregate Cancer Target Gene ID Gene name context+ P gene score CT ABL2 NM_001136000 V-abl Abelson murine leukemia viral oncogene homolog 2 > -0.03 0.31 √ ACAN NM_001135 Aggrecan -0.09 0.12 ACCN2 NM_001095 Amiloride-sensitive cation channel 2, neuronal > -0.01 0.26 ACER3 NM_018367 Alkaline ceramidase 3 -0.04 <0.1 ACVR2A NM_001616 Activin A receptor, type IIA -0.16 0.64 ADAM metallopeptidase with thrombospondin type 1 ADAMTS1 NM_006988 > -0.02 0.42 motif, 1 ADAM metallopeptidase with thrombospondin type 1 ADAMTS18 NM_199355 -0.17 0.64 motif, 18 ADAM metallopeptidase with thrombospondin type 1 ADAMTS5 NM_007038 -0.24 0.59 motif, 5 ADAMTSL1 NM_001040272 ADAMTS-like 1 -0.26 0.72 ADARB1 NM_001112 Adenosine deaminase, RNA-specific, B1 -0.28 0.62 AFAP1 NM_001134647 Actin filament associated protein 1 -0.09 0.61 AFTPH NM_001002243 Aftiphilin -0.16 0.49 AK3 NM_001199852 Adenylate kinase 3 > -0.02 0.6 AKAP7 NM_004842 A kinase (PRKA) anchor protein 7 -0.16 0.37 ANAPC16 NM_001242546 Anaphase promoting complex subunit 16 -0.1 0.66 ANK1 NM_000037 Ankyrin 1, erythrocytic > -0.03 0.46 ANKRD12 NM_001083625 Ankyrin repeat domain 12 > -0.03 0.31 ANKRD33B NM_001164440 Ankyrin repeat domain 33B -0.17 0.35 ANKRD43 NM_175873 Ankyrin repeat domain 43 -0.16 0.65 ANKRD44 NM_001195144 Ankyrin repeat domain 44 -0.17 0.49 ANKRD52 NM_173595 Ankyrin repeat domain 52 > -0.05 0.7 AP1S3 NM_001039569 Adaptor-related protein complex 1, sigma 3 subunit -0.26 0.76 Amyloid beta (A4) precursor protein-binding, family A, APBA1 NM_001163 -0.13 0.81 member 1 APLP2 NM_001142276 Amyloid beta (A4) precursor-like protein 2 -0.05 0.55 APOO NM_024122 Apolipoprotein O -0.32 0.41 ARID2 NM_152641 AT rich interactive domain 2 (ARID, RFX-like) -0.07 0.55 √ ARL3 NM_004311 ADP-ribosylation factor-like 3 > -0.03 0.51 ARRDC3 NM_020801 Arrestin domain containing 3 > -0.02 0.47 ATF7 NM_001130059 Activating transcription factor 7 > -0.01 0.26 ATG2B NM_018036 ATG2 autophagy related 2 homolog B (S. -

Systematic Analysis of Lysine Acetyltransferases
Systematic Analysis of Lysine Acetyltransferases Dissertation zur Erlangung des akademischen Grades Dr. rer. nat. vorgelegt der Fakultät für Biologie der Ludwig-Maximilians-Universität München Christian Feller München 2014 1. Gutachter: Prof. Dr. Peter B. Becker 2. Gutachter: Prof. Dr. Dirk Eick Dissertation eingereicht am: 16.09.2014 Mündliche Prüfung am: 28.10.2014 Eidesstattliche Erklärung Ich versichere hiermit an Eides statt, dass die vorgelegte Dissertation von mir selbstständig und ohne unerlaubte Hilfe angefertigt ist. München, den 15. September 2014 .................................... (Christian Feller) Erklärung Hiermit erkläre ich, dass die vorgelegte Arbeit an der LMU von Herrn Prof. Dr. Peter Becker betreut wurde. Hiermit erkläre ich, dass die Dissertation nicht ganz oder in Teilen einer anderen Prüfungskommission vorgelegt worden ist. Weiterhin habe ich weder an einem anderen Ort eine Promotion angestrebt noch angemeldet, noch versucht eine Doktorprüfung abzulegen. Die eigenen Leistungen für die in dieser kumulativen Dissertation enthaltenen Manuskripte sind in den Kapiteln 3.1.1, 3.2.1, 3.3.1 und 3.4.1 aufgelistet. München, den 15. September 2014 .................................... (Christian Feller) For my parents Table of contents 1 SUMMARY ................................................................................................................................. 11 2 INTRODUCTION ........................................................................................................................ 15 2.1 The -

Insights Into the Comparative Biological Roles of S. Cerevisiae Nucleoplasmin-Like Fkbps Fpr3 and Fpr4
Insights into the comparative biological roles of S. cerevisiae nucleoplasmin-like FKBPs Fpr3 and Fpr4 by Neda Savic B.Sc. Portland State University, 2012 A Dissertation Submitted in Partial Fulfillment of the Requirements for the Degree of DOCTOR OF PHILOSOPHY in the Department of Biochemistry and Microbiology © Neda Savic, 2019 University of Victoria All rights reserved. This dissertation may not be reproduced in whole or in part, by photocopy or other means, without the written permission of the author. ii Supervisory Committee Insights into the comparative biological roles of S. cerevisiae nucleoplasmin-like FKBPs Fpr3 and Fpr4 by Neda Savic B.Sc. Portland State University, 2012 Supervisory Committee Dr. Christopher J. Nelson, Supervisor Department of Biochemistry and Microbiology Dr. Juan Ausio, Departmental Member Department of Biochemistry and Microbiology Dr. Caren C. Helbing, Departmental Member Department of Biochemistry and Microbiology Dr. Peter C. Constabel, Outside Member Department of Biology iii Abstract The nucleoplasmin (NPM) family of acidic histone chaperones and the FK506-binding (FKBP) peptidyl proline isomerases are both linked to chromatin regulation. In vertebrates, NPM and FKBP domains are found on separate proteins. In fungi, NPM-like and FKBP domains are expressed as a single polypeptide in nucleoplasmin-like FKBP (NPL-FKBP) histone chaperones. Saccharomyces cerevisiae has two NPL-FKBPs: Fpr3 and Fpr4. These paralogs are 72% similar and are clearly derived from a common ancestral gene. This suggests that they may have redundant functions. Their retention over millions of years of evolution also implies that each must contribute non-redundantly to organism fitness. The redundant and separate biological functions of these chromatin regulators have not been studied. -

Functional Analysis of Expressed Sequence Tags from the Liver and Brain of Korean Jindo Dogs
BMB reports Functional analysis of expressed sequence tags from the liver and brain of Korean Jindo dogs Jae Young Kim#,1,3, Hye Sun Park#,1, Dajeong Lim1, Hong Chul Jang1, Hae Suk Park1, Kyung Tai Lee1, Jong Seok Kim2, Seok Il Oh2, Mu Sik Kweon3, Tae Hun Kim1 & Bong Hwan Choi1,* 1Division of Animal Genomics and Bioinformatics, National Institute of Animal Science, Rural Development Administration, Suwon 441-706, 2Korean Jindo and Domestic Animals Center, Jindo 539-800, 3Department of Genetic Engineering, Sungkyunkwan University, Suwon 440-746, Korea We generated 16,993 expressed sequence tags (ESTs) from dogs generally live in the same environment as their owner, two libraries containing full-length cDNAs from the brain and sharing living space and food. Second, dogs and humans share liver of the Korean Jindo dog. An additional 365,909 ESTs susceptibility to many diseases, including cancers, blindness, from other dog breeds were identified from the NCBI dbEST cataracts, deafness, epilepsy, heart disease, and genetic dis- database, and all ESTs were clustered into 28,514 consensus orders and often exhibit symptoms similar to humans (3). sequences using StackPack. We selected the 7,305 consensus Moreover, some diseases can be treated more easily in dogs sequences that could be assembled from at least five ESTs and than in humans (3). Finally, dogs often have a high level of estimated that 12,533 high-quality single nucleotide poly- health care, and the canine lifespan is more than five-fold shorter morphisms (SNPs) were present in 97,835 putative SNPs from than that of humans, making dogs excellent model organisms.