Functional Analysis of Penicillium Paxilli Genes Required for Biosynthesis of Paxilline
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
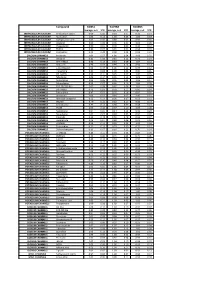
Biomol Average and SD Table S1.Xlsx
Compound GliNS1 G179NS G166NS average, n=5 STD average, n=3 STD average, n=3 STD INTRACELLULAR CALCIUM Antibiotic A-23187 0.00 0.00 0.10 0.07 0.15 0.14 INTRACELLULAR CALCIUM Ryanodine 1.04 0.14 1.03 0.03 1.03 0.03 INTRACELLULAR CALCIUM Cyclopiazonic acid 1.01 0.06 0.88 0.05 0.92 0.06 INTRACELLULAR CALCIUM Gingerol 1.00 0.06 0.91 0.01 1.01 0.06 INTRACELLULAR CALCIUM Thapsigargin 0.00 0.01 0.00 0.00 0.10 0.12 INTRACELLULAR CALCIUM TMB-8 0.89 0.07 0.91 0.05 0.94 0.03 INTRACELLULAR CALCIUM Dantrolene 0.91 0.08 0.98 0.05 0.94 0.01 CALCIUM CHANNELS Amiloride 1.01 0.07 1.01 0.04 1.03 0.05 CALCIUM CHANNELS Benzamil 0.83 0.08 0.83 0.12 0.96 0.04 CALCIUM CHANNELS BAY K-8644 0.93 0.13 0.93 0.09 1.07 0.14 CALCIUM CHANNELS Diltiazem 0.96 0.07 0.99 0.12 0.94 0.14 CALCIUM CHANNELS L-cis-Diltiazem 0.91 0.17 1.01 0.12 0.95 0.12 CALCIUM CHANNELS Flunarizine 0.85 0.08 1.00 0.06 0.85 0.05 CALCIUM CHANNELS FPL-64176 0.99 0.11 0.95 0.07 1.05 0.05 CALCIUM CHANNELS Nifedipine 1.06 0.17 0.95 0.12 1.03 0.09 CALCIUM CHANNELS Nimodipine 1.05 0.06 0.95 0.03 1.06 0.17 CALCIUM CHANNELS Nitrendipine 0.99 0.07 0.96 0.10 1.04 0.09 CALCIUM CHANNELS SDZ-202791 R(-) 1.01 0.08 0.92 0.06 1.01 0.08 CALCIUM CHANNELS SKF-96365 0.73 0.05 0.70 0.11 0.69 0.04 CALCIUM CHANNELS Tetrandrine 0.47 0.07 0.76 0.16 0.87 0.20 CALCIUM CHANNELS Verapamil 1.01 0.02 0.89 0.07 1.06 0.20 CALCIUM CHANNELS Methoxy Verapamil 0.93 0.14 0.96 0.07 0.93 0.13 CALCIUM CHANNELS Bepridil 0.70 0.16 0.92 0.15 0.84 0.14 CALCIUM CHANNELS Amiodarone 0.32 0.12 0.58 0.07 0.48 0.23 CALCIUM CHANNELS YS035 1.00 0.16 -

Big Conductance Calcium-Activated Potassium Channel Openers Control Spasticity Without Sedation
British Journal of British Journal of Pharmacology (2017) 174 2662–2681 2662 BJP Pharmacology RESEARCH PAPER Big conductance calcium-activated potassium channel openers control spasticity without sedation Correspondence Professor David Baker, Neuroimmunology Unit, Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, 4 Newark Street, Whitechapel, London E1 4AT, UK. E-mail: [email protected] Received 5 January 2017; Revised 27 April 2017; Accepted 17 May 2017 David Baker1,2,* ,GarethPryce1,2,*, Cristina Visintin2,3,SofiaSisay1, Alexander I Bondarenko4,5, WSVanessaHo6, Samuel J Jackson1, Thomas E Williams1, Sarah Al-Izki1, Ioanna Sevastou3, Masahiro Okuyama3, Wolfgang F Graier4, Lesley A Stevenson6,CarolynTanner7,RuthRoss7, Roger G Pertwee7, Christopher M Henstridge8,AndrewJIrving8, Jesse Schulman9,KeithPowell9,MarkDBaker1, Gavin Giovannoni1,2 and David L Selwood3,* 1Neuroimmunology Unit, Blizard Institute, Barts and the London School of Medicine and Dentistry, Queen Mary University of London, London, UK, 2Department of Neuroinflammation, UCL Institute of Neurology, University College London, London, UK, 3Department of Medicinal Chemistry, UCL Wolfson Institute for Biomedical Research, University College London, London, UK, 4Institute of Molecular Biology and Biochemistry, Medical University of Graz, Graz, Austria, 5A.A. Bogomoletz Institute of Physiology, Kiev, Ukraine, 6Vascular Biology Research Centre. St. George’s, University of London, London, UK, 7Department of Biomedical Sciences, -

Hypercholesterolemia Effect on Potassium Channels
We are IntechOpen, the world’s leading publisher of Open Access books Built by scientists, for scientists 5,400 134,000 165M Open access books available International authors and editors Downloads Our authors are among the 154 TOP 1% 12.2% Countries delivered to most cited scientists Contributors from top 500 universities Selection of our books indexed in the Book Citation Index in Web of Science™ Core Collection (BKCI) Interested in publishing with us? Contact [email protected] Numbers displayed above are based on latest data collected. For more information visit www.intechopen.com Chapter 5 Hypercholesterolemia Effect on Potassium Channels Anna N. Bukiya and Avia Rosenhouse-Dantsker Additional information is available at the end of the chapter http://dx.doi.org/10.5772/59761 1. Introduction Cholesterol is a major lipid component of the plasma membrane in mammalian cells consti‐ tuting up to 45 mol % with respect to other lipids [1, 2]. Yet, even a limited increase in blood and/or tissue cholesterol of up to 2-3 fold above the physiological level is cytotoxic [1-3] and is associated with the development of cardiovascular disease [4-6]. The underlying source for the effect of cholesterol on cellular functions is its ability to alter the function of multiple membrane proteins including ion channels (see, for example, reviews [7-9]). In recent years, high cholesterol diet has been shown to affect the function of multiple ion channels. In this chapter we focus on the effect of dietary-induced increase in blood and tissue cholesterol levels on potassium channels. Potassium channels are among the largest and most complex types of ion channels. -

Correlation of Fecal Ergovaline, Lolitrem B, and Their Metabolites in Steers Fed Endophyte Infected Perennial Ryegrass Straw
AN ABSTRACT OF THE THESIS OF Lia D. Murty for the degree of Master of Science in Pharmacy presented on November 21, 2012. Title: Correlation of Fecal Ergovaline, Lolitrem B, and their Metabolites in Steers Fed Endophyte Infected Perennial Ryegrass Straw Abstract approved: A. Morrie Craig Perennial ryegrass (PRG, Lolium perenne) is a hardy cool-season grass that is infected with the endophytic fungus Neotyphodium lolii, which enables the plant to be insect repellant and drought resistant, lowering the use of insecticides and fertilizers. However, this fungus produces the compound lolitrem B (LB, m/z 686.4) which causes the tremorgenic neurotoxicity syndrome ‘ryegrass staggers’ in livestock consuming forage which contains <2000 ppb LB. Ergovaline (EV, m/z 534) is a vasoconstrictor normally associated with tall fescue (Festuca arudinacea), but has also been found in endophyte- infected PRG. Past research has shown a strong linear correlation between levels of LB and EV in PRG. The purpose of this study was to examine the linear relationship between EV and LB in feces and determine common metabolites. To accomplish this, four groups of steers (n=6/group) consumed endophyte- infected PRG over 70 days consumed the following averages of LB and EV: group I 2254ppb LB/633 ppb EV; group II 1554ppb LB/ 373ppb EV, group III 1011ppb LB/259ppb EV, and group IV 246ppb LB/<100ppb EV. Group I in week 4 was inadvertently given a washout period at which time the steers consumed the amount of LB and EV given to group IV (control). Both feed and feces samples were extracted using difference solid phase extraction methods and quantified by HPLC-fluorescence for LB and EV. -

WO 2016/123419 Al 4 August 2016 (04.08.2016) P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization I International Bureau (10) International Publication Number (43) International Publication Date WO 2016/123419 Al 4 August 2016 (04.08.2016) P O P C T (51) International Patent Classification: AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, C12Q 1/68 (2006.01) C07H 21/02 (2006.01) BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, (21) International Application Number: HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, PCT/US2016/015503 KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, (22) International Filing Date: MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, 29 January 2016 (29.01 .2016) PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, (25) Filing Language: English TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. (26) Publication Language: English (84) Designated States (unless otherwise indicated, for every (30) Priority Data: kind of regional protection available): ARIPO (BW, GH, 62/1 10,050 30 January 2015 (30.01.2015) US GM, KE, LR, LS, MW, MZ, NA, RW, SD, SL, ST, SZ, TZ, UG, ZM, ZW), Eurasian (AM, AZ, BY, KG, KZ, RU, (71) Applicant: PRESIDENT AND FELLOWS OF HAR¬ TJ, TM), European (AL, AT, BE, BG, CH, CY, CZ, DE, VARD COLLEGE [US/US]; 17 Quincy Street, Cam DK, EE, ES, FI, FR, GB, GR, HR, HU, IE, IS, IT, LT, LU, bridge, MA 02138 (US). -

The Role of BK Channels in Antiseizure Action of the CB1 Receptor Agonist ACEA in Maximal Electroshock and Pentylenetetrazole Models of Seizure in Mice
Iranian Journal of Pharmaceutical Research (2017), 16 (2): 640-647 Copyright © 2017 by School of Pharmacy Received: Aug. 2016 Shaheed Beheshti University of Medical Sciences and Health Services Accepted: Feb. 2017 Original Article The Role of BK Channels in Antiseizure Action of the CB1 Receptor Agonist ACEA in Maximal Electroshock and Pentylenetetrazole Models of Seizure in Mice Sina Asaadia, Mohammad Jahanbakhshia, Mahmoud Lotfiniab and Nima Naderia,c* aNeuroscience Research Center, Shahid Beheshti University of Medical Sciences, Tehran, Iran. bSchool of Medicine, Shahid Beheshti University of Medical Sciences, Tehran, Iran. cDepartment of Pharmacology and Toxicology, School of Pharmacy, Shahid Beheshti University of Medical Sciences, Tehran, Iran. Abstract The anticonvulsant effect of cannabinoid compound has been shown in various models of seizure. On the other hand, there are controversial findings about the role of large conductance calcium-activated potassium (BK) channels in the pathogenesis of epilepsy. Also, there is no data regarding the effect of co-administration of cannabinoid type 1 (CB1) receptor agonists and BK channels antagonists in the acute models of seizure in mice. In this study, the effect of arachidonyl-2′-chloroethylamide (ACEA), a CB1 receptor agonist, and a BK channel antagonist, paxilline, either alone or in combination was investigated. Both pentylenetetrazole (PTZ) and maximal electroshock (MES) acute models of seizure were used to evaluate the protective effects of drugs. Mice were randomly selected in different groups: (i) control group; (ii) groups that received different doses of either paxilline or ACEA; and (iii) groups that received combinations of ACEA and paxillin at different doses. In MES model, prevention of hindlimb tonic extension (HLTE) was considered as protective effect. -

Fifth Annual Neuroscience, Behavior and Health Research Forum
FIFTH ANNUAL NEUROSCIENCE, BEHAVIOR AND HEALTH RESEARCH FORUM The University of Vermont Dudley H. Davis Center Livak Ballroom / Mansfield Room January 23 - 24, 2015 Platform Talks and Poster Abstracts Sponsored by: Society for Neuroscience Society for Neuroscience Vermont Chapter UVM Neuroscience, Behavior and Health Initiative UVM Neuroscience Graduate Program Neuroscience COBRE MBF Bioscience Med Associates / Catamount Research Platform Talk 1 Brain-derived neurotrophic factor: a novel regulator of cardiovascular function Benedek Erdos Department of Pharmacology, University of Vermont College of Medicine, Burlington, VT Brain-derived neurotrophic factor (BDNF) is a member of the neurotrophin family and has a key role in regulating neuronal development and survival. In addition, increasing evidence indicates that neuronal activity-dependent production and release of BDNF provokes both short- term and long-term changes in synaptic function and that BDNF may also act as a neurotransmitter. The paraventricular nucleus of the hypothalamus (PVN) plays a central role in neural control of cardiovascular function, and BDNF synthesis in the PVN has been shown to increase in response to hypertensive stimuli including stress and hyperosmolarity. However, it is unclear whether BDNF, acting within the PVN, contributes to elevations in blood pressure. Using radiotelemetric blood pressure monitoring in Sprague-Dawley rats, we have established that 1) viral vector-mediated overexpression of BDNF in the PVN induces significant increases in mean arterial pressure; heart rate and indices of sympathetic activity; 2) acute microinjection of BDNF into the PVN elicits rapid elevations in blood pressure and heart rate; 3) both acute and long-term effects of BDNF are mediated at least in part by changes in the brain renin angiotensin system; 4) chronically elevated BDNF in the PVN upregulates catecholamine biosynthesizing enzymes in certain nuclei of the brainstem. -

Phytocannabinoids-And-Epilepsy.Pdf
Journal of Clinical Pharmacy and Therapeutics, 2014 doi: 10.1111/jcpt.12235 Review Article Phytocannabinoids and epilepsy R. G. dos Santos* PhD, J. E. C. Hallak*† MD PhD, J. P. Leite* MD PhD, A. W. Zuardi*† MD PhD and J. A. S. Crippa*† MD PhD *Department of Neuroscience and Behavior, Ribeir~ao Preto Medical School, University of S~ao Paulo, and †National Institute for Translational Medicine (INCT-TM), CNPq, Ribeir~ao Preto, Brazil Received 4 September 2014, Accepted 6 November 2014 Keywords: cannabidiol, cannabinoids, delta-9-tetrahydrocannabinol, epilepsy, seizure, treatment nabivarin (THCV), cannabidivarin (CBDV), delta-8-tetrahydrocan- SUMMARY nabinol (delta-8-THC), cannabinol (CBN) and especially CBD have – – What is known and objective: Antiepileptic drugs often produce anticonvulsant effects.13 17,23 29 Considering that currently avail- serious adverse effects, and many patients do not respond to able anti-epileptic drugs are not efficient for many patients and them properly. Phytocannabinoids produce anticonvulsant that these drugs often produce several side effects, there is a clear effects in preclinical and preliminary human studies, and appear need to explore other compounds with higher efficacy and lower to produce fewer adverse effects than available antiepileptic toxicity.23,24 Therefore, the present text presents a review of the drugs. The present review summarizes studies on the anticon- studies on the anticonvulsant effects of phytocannabinoids. vulsant properties of phytocannabinoids. Methods: Literature search using the PubMed database to METHODS identify studies on phytocannabinoids and epilepsy. Results and discussion: Preclinical studies suggest that phytoc- A literature search was performed in the PubMed database using annabinoids, especially cannabidiol and cannabidivarin, have the words ‘cannabis’, ‘marijuana’, ‘cannabinoids’, ‘tetrahydrocan- potent anticonvulsant effects which are mediated by the endoc- nabinol’, ‘THC’, ‘cannabidiol’, ‘CBD’, ‘cannabidivarin’, ‘CBDV’, annabinoid system. -

A Comprehensive Map of Disease Networks and Molecular Drug Discoveries for Glaucoma Haixin Wang1,2,3, Yanhui Deng1, Ling Wan4 & Lulin Huang1,2,3 ✉
www.nature.com/scientificreports OPEN A comprehensive map of disease networks and molecular drug discoveries for glaucoma Haixin Wang1,2,3, Yanhui Deng1, Ling Wan4 & Lulin Huang1,2,3 ✉ Glaucoma is the leading cause of irreversible blindness worldwide. The molecular etiology of glaucoma is complex and unclear. At present, there are few drugs available for glaucoma treatment. The aim of the present study was to perform a systematic analysis of glaucoma candidate drugs/chemicals based on glaucoma genes, including genetic factors and diferentially expressed (DE) genes. In total, 401 genes from the genetic databases and 1656 genes from the DE gene analysis were included in further analyses. In terms of glaucoma-related genetic factors, 54 pathways were signifcantly enriched (FDR < 0.05), and 96 pathways for DE genes were signifcantly enriched (FDR < 0.05). A search of the PheWAS database for diseases associated with glaucoma-related genes returned 1,289 diseases, and a search for diseases associated with DE glaucoma-related genes returned 1,356 diseases. Cardiovascular diseases, neurodegenerative diseases, cancer, and ophthalmic diseases were highly related to glaucoma genes. A search of the DGIdb, KEGG, and CLUE databases revealed a set of drugs/chemicals targeting glaucoma genes. A subsequent analysis of the electronic medical records (EMRs) of 136,128 patients treated in Sichuan Provincial People’s Hospital for candidate drug usage and the onset of glaucoma revealed nine candidate drugs. Among these drugs, individuals treated with nicardipine had the lowest incidence of glaucoma. Taken together with the information from the drug databases, the 40 most likely candidate drugs for glaucoma treatment were highlighted. -

Natural Modulators of Large-Conductance Calcium
Antonio Nardi1 Vincenzo Calderone2 Natural Modulators of Large-Conductance Silvio Chericoni3 Ivano Morelli3 Calcium-Activated Potassium Channels Review Abstract wards identifying new BK-modulating agents is proceeding with great impetus and is giving an ever-increasing number of Large-conductance calcium-activated potassium channels, also new molecules. Among these, also a handsome number of natur- known as BK or Maxi-K channels, occur in many types of cell, in- al BK-modulator compounds, belonging to different structural cluding neurons and myocytes, where they play an essential role classes, has appeared in the literature. The goal of this paper is in the regulation of cell excitability and function. These proper- to provide a possible simple classification of the broad structural ties open a possible role for BK-activators also called BK-open- heterogeneity of the natural BK-activating agents terpenes, phe- ers) and/or BK-blockers as effective therapeutic agents for differ- nols, flavonoids) and blockers alkaloids and peptides), and a ent neurological, urological, respiratory and cardiovascular dis- concise overview of their chemical and pharmacological proper- eases. The synthetic benzimidazolone derivatives NS004 and ties as well as potential therapeutic applications. NS1619 are the pioneer BK-activators and have represented the reference models which led to the design of several novel and Key words heterogeneous synthetic BK-openers, while very few synthetic Natural products ´ potassium channels ´ large-conductance cal- BK-blockers have been reported. Even today, the research to- cium-activated BK channels ´ BK-activators ´ BK-blockers 885 Introduction tracellular Ca2+ and membrane depolarisation, promoting a mas- sive outward flow of K+ ions and leading to a membrane hyper- Among the different factors exerting an influence on the activity polarisation, i.e., to a stabilisation of the cell [1]. -

Www .Alfa.Com
Bio 2013-14 Alfa Aesar North America Alfa Aesar Korea Uni-Onward (International Sales Headquarters) 101-3701, Lotte Castle President 3F-2 93 Wenhau 1st Rd, Sec 1, 26 Parkridge Road O-Dong Linkou Shiang 244, Taipei County Ward Hill, MA 01835 USA 467, Gongduk-Dong, Mapo-Gu Taiwan Tel: 1-800-343-0660 or 1-978-521-6300 Seoul, 121-805, Korea Tel: 886-2-2600-0611 Fax: 1-978-521-6350 Tel: +82-2-3140-6000 Fax: 886-2-2600-0654 Email: [email protected] Fax: +82-2-3140-6002 Email: [email protected] Email: [email protected] Alfa Aesar United Kingdom Echo Chemical Co. Ltd Shore Road Alfa Aesar India 16, Gongyeh Rd, Lu-Chu Li Port of Heysham Industrial Park (Johnson Matthey Chemicals India Toufen, 351, Miaoli Heysham LA3 2XY Pvt. Ltd.) Taiwan England Kandlakoya Village Tel: 866-37-629988 Bio Chemicals for Life Tel: 0800-801812 or +44 (0)1524 850506 Medchal Mandal Email: [email protected] www.alfa.com Fax: +44 (0)1524 850608 R R District Email: [email protected] Hyderabad - 501401 Andhra Pradesh, India Including: Alfa Aesar Germany Tel: +91 40 6730 1234 Postbox 11 07 65 Fax: +91 40 6730 1230 Amino Acids and Derivatives 76057 Karlsruhe Email: [email protected] Buffers Germany Tel: 800 4566 4566 or Distributed By: Click Chemistry Reagents +49 (0)721 84007 280 Electrophoresis Reagents Fax: +49 (0)721 84007 300 Hydrus Chemical Inc. Email: [email protected] Uchikanda 3-Chome, Chiyoda-Ku Signal Transduction Reagents Tokyo 101-0047 Western Blot and ELISA Reagents Alfa Aesar France Japan 2 allée d’Oslo Tel: 03(3258)5031 ...and much more 67300 Schiltigheim Fax: 03(3258)6535 France Email: [email protected] Tel: 0800 03 51 47 or +33 (0)3 8862 2690 Fax: 0800 10 20 67 or OOO “REAKOR” +33 (0)3 8862 6864 Nagorny Proezd, 7 Email: [email protected] 117 105 Moscow Russia Alfa Aesar China Tel: +7 495 640 3427 Room 1509 Fax: +7 495 640 3427 ext 6 CBD International Building Email: [email protected] No. -

Low Chloride Conductance Myotonia - in Vitro Investigations on Muscle Stiffness and the Warm-Up Phenomenon
Ulm University Institute of Applied Physiology Prof. Dr. Dr. h.c. Frank Lehmann-Horn Low chloride conductance myotonia - in vitro investigations on muscle stiffness and the warm-up phenomenon Dissertation Applying for the Degree of Doctor of human biology (Dr. biol. hum.) Faculty of Medicine, Ulm University Submitted by Sunisa Chaiklieng From Phatthalung, Thailand 2007 Amtierender Dekan: Prof. Dr. Klaus-Michael Debatin 1. Berichterstatter: Prof. Dr. Dr. h.c. Frank Lehmann-Horn 2. Berichterstatter: Prof. Dr. Holger Lerche Tag der Promotion: 18.12.2007 I TABLE OF CONTENTS ABBREVIATIONS 1 INTRODUCTION 1 1.1 MYOTONIA - HYPEREXCITABILITY OF SKELETAL MUSCLE....................................... 1 1.2 LOW CHLORIDE CONDUCTANCE MYOTONIA ............................................................ 1 1.2.1 THOMSEN’S AND BECKER’S MYOTONIA ................................................................... 1 1.2.2 PATHOPHYSIOLOGICAL BACKGROUND ..................................................................... 3 1.2.3 WARM-UP PHENOMENON.......................................................................................... 3 1.3 PHYSIOLOGICAL AND MICROENVIRONMENTAL MODIFYING FACTORS ..................... 4 1.4 GENETICS ................................................................................................................. 5 1.5 DIFFERENT EXPRESSION PATTERNS OF SARCOLEMMAL AND T-TUBULAR MEMBRANE PROTEINS.............................................................................................. 6 - 1.6 PHARMACOLOGICALLY INDUCED LOW