Bkca Channels As Targets for Cardioprotection
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Effect of Hypercholesterolaemia on Voltage-Operated Calcium Channel Currents in Rabbit Arterial Smooth Muscle Cells
Journal of Human Hypertension (1999) 13, 849–853 1999 Stockton Press. All rights reserved 0950-9240/99 $15.00 http://www.stockton-press.co.uk/jhh Effect of hypercholesterolaemia on voltage-operated calcium channel currents in rabbit arterial smooth muscle cells GF Clunn, S Wijetunge and AD Hughes Clinical Pharmacology, NHLI, St. Mary’s Hospital, Imperial College of Science, Technology and Medicine, South Wharf Road, London, W2 1NY, UK Cholesterol is a major component of cell membranes capacitance was also greater in NZ cells. Consequently, and influences membrane fluidity. Watanabe heritable there was no significant difference in current density hyperpercholesterolaemic rabbits (WHHL) possess between NZ and WHHL cells either in the absence of defective receptors for low density lipoprotein leading drug or in the presence of the calcium channel agonist to increased plasma cholesterol, accumulation of chol- (+)202 791. Current voltage-relationships, kinetics of esterol in the arterial wall and atherosclerosis. In this fast inactivation and steady-state inactivation of IBa also study calcium channel currents (IBa) were compared did not differ significantly between WHHL and NZ. These using conventional whole cell voltage clamp techniques findings suggest that hypercholesterolaemia in WHHL in ear artery cells isolated from control New Zealand has no direct effect on calcium channel current density White rabbits (NZ) with those from WHHL. IBa were larger or voltage-modulation in arterial smooth muscle cells. in cells isolated from NZ than from WHHL, however cell Keywords: calcium channel; cholesterol; vascular smooth muscle; Watanabe hypercholesterolaemic rabbit Introduction atic cholesterol toxicity or other organ damage which develops in cholesterol-fed rabbits.15 WHHL Cholesterol is a major component of cell membranes 1,2 has therefore been proposed to be a model of human and influences membrane structure and fluidity. -

De Novo BK Channel Variant Causes Epilepsy by Affecting Voltage Gating but Not Ca2+ Sensitivity
European Journal of Human Genetics (2018) 26:220–229 https://doi.org/10.1038/s41431-017-0073-3 ARTICLE De novo BK channel variant causes epilepsy by affecting voltage gating but not Ca2+ sensitivity 1 2 3,4 5 6 Xia Li ● Sibylle Poschmann ● Qiuyun Chen ● Walid Fazeli ● Nelly Jouayed Oundjian ● 7 8 9 9,10 Francesca M. Snoeijen-Schouwenaars ● Oliver Fricke ● Erik-Jan Kamsteeg ● Marjolein Willemsen ● Qing Kenneth Wang1,3,4 Received: 18 August 2017 / Revised: 6 November 2017 / Accepted: 23 November 2017 / Published online: 12 January 2018 © European Society of Human Genetics 2018 Abstract Epilepsy is one of the most common neurological diseases and it causes profound morbidity and mortality. We identified the first de novo variant in KCNMA1 (c.2984 A > G (p.(N995S)))—encoding the BK channel—that causes epilepsy, but not paroxysmal dyskinesia, in two independent families. The c.2984 A > G (p.(N995S)) variant markedly increased the macroscopic potassium current by increasing both the channel open probability and channel open dwell time. The c.2984 A > G (p.(N995S)) variant did not affect the calcium sensitivity of the channel. We also identified three other variants of fi > > > 1234567890 unknown signi cance (c.1554 G T (p.(K518N)), c.1967A C (p.(E656A)), and c.3476 A G (p.(N1159S))) in three separate patients with divergent epileptic phenotypes. However, these variants did not affect the BK potassium current, and are therefore unlikely to be disease-causing. These results demonstrate that BK channel variants can cause epilepsy without paroxysmal dyskinesia. The underlying molecular mechanism can be increased activation of the BK channel by increased sensitivity to the voltage-dependent activation without affecting the sensitivity to the calcium-dependent activation. -

Holocene Environmental Changes Disclosed from Anoxic Fjord Sediments by Biomarkers and Their Radiocarbon Content
GEOLOGICA ULTRAIECTINA Mededelingen van de Faculteit Geowetenschappen Universiteit Utrecht No. 227 Holocene environmental changes disclosed from anoxic fjord sediments by biomarkers and their radiocarbon content Rienk H. Smittenberg Holocene environmental changes disclosed from anoxic fjord sediments by biomarkers and their radiocarbon content Holocene milieuveranderingen gereconstrueerd uit anoxische fjord sedimenten middels biomarkers en hun radio-aktief koolstof gehalte (met een samenvatting in het Nederlands) Proefschrift ter verkrijging van de graad van doctor aan de Universiteit Utrecht op gezag van de Rector Magnificus, Prof. Dr. W.H. Gispen, ingevolge het besluit van het College voor Promoties in het openbaar te verdedigen op maandag 15 september 2003 des middags te 4.15 uur door Rienk Hajo Smittenberg geboren op 23 maart 1973 te Eck en Wiel Promotor: Prof. Dr. J.W. de Leeuw Department of Geochemistry Utrecht University Utrecht, The Netherlands Copromotores: Dr. Ir. J.S. Sinninghe Damsté Department of Geochemistry Utrecht University Utrecht, The Netherlands Dr. S. Schouten Department of Marine Biogeochemistry and Toxicology Royal Netherlands Institute of Sea Research Texel, The Netherlands The research described in this thesis was carried out at the Department of Biogeochemistry and Toxicology of the Royal Netherlands Institute of Sea Research, P.O. Box 59, 1790 AB Den Burg, The Netherlands. The investigations were supported by the Research Council for Earth and Life Science (ALW) with the financial support from the Netherlands -
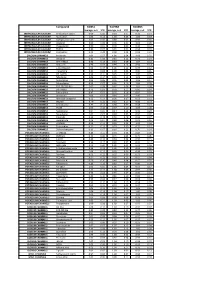
Biomol Average and SD Table S1.Xlsx
Compound GliNS1 G179NS G166NS average, n=5 STD average, n=3 STD average, n=3 STD INTRACELLULAR CALCIUM Antibiotic A-23187 0.00 0.00 0.10 0.07 0.15 0.14 INTRACELLULAR CALCIUM Ryanodine 1.04 0.14 1.03 0.03 1.03 0.03 INTRACELLULAR CALCIUM Cyclopiazonic acid 1.01 0.06 0.88 0.05 0.92 0.06 INTRACELLULAR CALCIUM Gingerol 1.00 0.06 0.91 0.01 1.01 0.06 INTRACELLULAR CALCIUM Thapsigargin 0.00 0.01 0.00 0.00 0.10 0.12 INTRACELLULAR CALCIUM TMB-8 0.89 0.07 0.91 0.05 0.94 0.03 INTRACELLULAR CALCIUM Dantrolene 0.91 0.08 0.98 0.05 0.94 0.01 CALCIUM CHANNELS Amiloride 1.01 0.07 1.01 0.04 1.03 0.05 CALCIUM CHANNELS Benzamil 0.83 0.08 0.83 0.12 0.96 0.04 CALCIUM CHANNELS BAY K-8644 0.93 0.13 0.93 0.09 1.07 0.14 CALCIUM CHANNELS Diltiazem 0.96 0.07 0.99 0.12 0.94 0.14 CALCIUM CHANNELS L-cis-Diltiazem 0.91 0.17 1.01 0.12 0.95 0.12 CALCIUM CHANNELS Flunarizine 0.85 0.08 1.00 0.06 0.85 0.05 CALCIUM CHANNELS FPL-64176 0.99 0.11 0.95 0.07 1.05 0.05 CALCIUM CHANNELS Nifedipine 1.06 0.17 0.95 0.12 1.03 0.09 CALCIUM CHANNELS Nimodipine 1.05 0.06 0.95 0.03 1.06 0.17 CALCIUM CHANNELS Nitrendipine 0.99 0.07 0.96 0.10 1.04 0.09 CALCIUM CHANNELS SDZ-202791 R(-) 1.01 0.08 0.92 0.06 1.01 0.08 CALCIUM CHANNELS SKF-96365 0.73 0.05 0.70 0.11 0.69 0.04 CALCIUM CHANNELS Tetrandrine 0.47 0.07 0.76 0.16 0.87 0.20 CALCIUM CHANNELS Verapamil 1.01 0.02 0.89 0.07 1.06 0.20 CALCIUM CHANNELS Methoxy Verapamil 0.93 0.14 0.96 0.07 0.93 0.13 CALCIUM CHANNELS Bepridil 0.70 0.16 0.92 0.15 0.84 0.14 CALCIUM CHANNELS Amiodarone 0.32 0.12 0.58 0.07 0.48 0.23 CALCIUM CHANNELS YS035 1.00 0.16 -

Application of Biomarker Compounds As Tracers for Sources and Fates of Natural and Anthropogenic Organic Matter in Tile Environment
AN ABSTRACT OF THE DISSERTATION OF Daniel R. Oros for the degree of Doctor of Philosophy in Environmental Sciences presented on September 24. 1999. Title: Application of Biomarker Compoundsas Tracers for Sources and Fates of Natural and Anthropogenic Organic Matter in the Environment. Redacted for Privacy Abstract approved: Bernd R.T. Simoneit Determination of the source and fate of natural (higher plant lipids, marine lipids, etc.) and anthropogenically (e.g., petroleum, coal emissions) derived hydrocarbons and oxygenated compounds in the environment was accomplished using gas chromatography (GC) and gas chromatography-mass spectrometry (GC- MS) to characterize or identify molecular biomarkers to be utilized as tracers. The distributions and abundances of biomarkers such as straight chain homologous series (e.g., n-alkanes, n-alkanoic acids, n-alkan-2-ones, n-alkanols, etc.) and cyclic terpenoid compounds (e.g., sesquiterpenoids, diterpenoids, steroids, triterpenoids) were identified in epicuticular waxes from conifers of western North America (natural emissions). These biomarkers and their thermal alteration derivativeswere also identified in smoke emissions from known vegetation sources (e.g., conifers, deciduous trees and grasses) and were then applied as tracers in soils, soils that contained wildfire residues and soillriver mud washout after wildfire burning. Where possible, the reaction pathways of transformation from the parentprecursor compounds to intermediate and final alteration products were determined from GC- MS data. In addition, molecular tracer analysis was applied to air, water and sediment samples collected from a lacustrine setting (Crater Lake, OR) in order to determine the identities, levels and fates of anthropogenic (i.e., petroleum hydrocarbon contamination from boating and related activities) hydrocarbons ina pristine organic matter sink. -

Emerging Roles for Multifunctional Ion Channel Auxiliary Subunits in Cancer T ⁎ Alexander S
Cell Calcium 80 (2019) 125–140 Contents lists available at ScienceDirect Cell Calcium journal homepage: www.elsevier.com/locate/ceca Emerging roles for multifunctional ion channel auxiliary subunits in cancer T ⁎ Alexander S. Hawortha,b, William J. Brackenburya,b, a Department of Biology, University of York, Heslington, York, YO10 5DD, UK b York Biomedical Research Institute, University of York, Heslington, York, YO10 5DD, UK ARTICLE INFO ABSTRACT Keywords: Several superfamilies of plasma membrane channels which regulate transmembrane ion flux have also been Auxiliary subunit shown to regulate a multitude of cellular processes, including proliferation and migration. Ion channels are Cancer typically multimeric complexes consisting of conducting subunits and auxiliary, non-conducting subunits. Calcium channel Auxiliary subunits modulate the function of conducting subunits and have putative non-conducting roles, further Chloride channel expanding the repertoire of cellular processes governed by ion channel complexes to processes such as trans- Potassium channel cellular adhesion and gene transcription. Given this expansive influence of ion channels on cellular behaviour it Sodium channel is perhaps no surprise that aberrant ion channel expression is a common occurrence in cancer. This review will − focus on the conducting and non-conducting roles of the auxiliary subunits of various Ca2+,K+,Na+ and Cl channels and the burgeoning evidence linking such auxiliary subunits to cancer. Several subunits are upregu- lated (e.g. Cavβ,Cavγ) and downregulated (e.g. Kvβ) in cancer, while other subunits have been functionally implicated as oncogenes (e.g. Navβ1,Cavα2δ1) and tumour suppressor genes (e.g. CLCA2, KCNE2, BKγ1) based on in vivo studies. The strengthening link between ion channel auxiliary subunits and cancer has exposed these subunits as potential biomarkers and therapeutic targets. -

Chemical Synthesis, Proper Folding, Nav Channel Selectivity Profile And
toxins Article Chemical Synthesis, Proper Folding, Nav Channel Selectivity Profile and Analgesic Properties of the Spider Peptide Phlotoxin 1 1, 2,3, 4,5, 1 Sébastien Nicolas y, Claude Zoukimian y, Frank Bosmans y,Jérôme Montnach , Sylvie Diochot 6, Eva Cuypers 5, Stephan De Waard 1,Rémy Béroud 2, Dietrich Mebs 7 , David Craik 8, Didier Boturyn 3 , Michel Lazdunski 6, Jan Tytgat 5 and Michel De Waard 1,2,* 1 Institut du Thorax, Inserm UMR 1087/CNRS UMR 6291, LabEx “Ion Channels, Science & Therapeutics”, F-44007 Nantes, France; [email protected] (S.N.); [email protected] (J.M.); [email protected] (S.D.W.) 2 Smartox Biotechnology, 6 rue des Platanes, F-38120 Saint-Egrève, France; [email protected] (C.Z.); [email protected] (R.B.) 3 Department of Molecular Chemistry, Univ. Grenoble Alpes, CNRS, 570 rue de la chimie, CS 40700, 38000 Grenoble, France; [email protected] 4 Faculty of Medicine and Health Sciences, Department of Basic and Applied Medical Sciences, 9000 Gent, Belgium; [email protected] 5 Toxicology and Pharmacology, University of Leuven, Campus Gasthuisberg, P.O. Box 922, Herestraat 49, 3000 Leuven, Belgium; [email protected] (E.C.); [email protected] (J.T.) 6 Université Côte d’Azur, CNRS UMR7275, Institut de Pharmacologie Moléculaire et Cellulaire, 660 route des lucioles, 6560 Valbonne, France; [email protected] (S.D.); [email protected] (M.L.) 7 Institute of Legal Medicine, University of Frankfurt, Kennedyallee 104, 60488 Frankfurt, Germany; [email protected] 8 Institute for Molecular Bioscience, University of Queensland, Brisbane 4072, Australia; [email protected] * Correspondence: [email protected]; Tel.: +33-228-080-076 Contributed equally to this work. -

Iberiotoxin-Induced Block of Kca Channels Induces Dihydropyridine
JPET Fast Forward. Published on January 24, 2003 as DOI: 10.1124/jpet.102.046102 JPET FastThis articleForward. has not Published been copyedited on and January formatted. 24,The final2003 version as DOI:10.1124/jpet.102.046102 may differ from this version. JPET/2002/46102 Iberiotoxin-induced Block of KCa Channels Induces Dihydropyridine Sensitivity of ACh Release from Mammalian Motor Nerve Terminals Downloaded from Michael T. Flink and William D. Atchison Department of Pharmacology & Toxicology jpet.aspetjournals.org Michigan State University E. Lansing, MI 48824 Address all correspondence including reprint requests to: at ASPET Journals on September 23, 2021 Dr. Bill Atchison Dept. Pharmacology & Toxicology Michigan State University B-331 Life Sciences Bldg. East Lansing, MI 48824-1317 Phone: (517) 353-4947 Fax: (517) 432-1341 Email: [email protected] 1 Copyright 2003 by the American Society for Pharmacology and Experimental Therapeutics. JPET Fast Forward. Published on January 24, 2003 as DOI: 10.1124/jpet.102.046102 This article has not been copyedited and formatted. The final version may differ from this version. JPET/2002/46102 Running Title: KCa Channel Block and DHP-sensitivity of ACh Release at NMJ (59 spaces) Document Statistics: Text Pages: 15 Tables: 1 Figures: 5 References: 42 Downloaded from Abstract: 249 words Introduction: 760 words Discussion: 1241 jpet.aspetjournals.org at ASPET Journals on September 23, 2021 LIST OF ABBREVIATIONS: ACh, acetylcholine; BSA, bovine serum albumin; Cav, voltage-activated calcium channel; DAP, 3,4 diaminopyridine; DHP, dihydropyridine;EPP, end-plate potential; HEPES, N-2- hydroxyethylpiperazine-N-2-ethanesulfonic acid ; KCa, calcium-activated potassium; MEPP, miniature end-plate potential 2 JPET Fast Forward. -

Interplay Between Gating and Block of Ligand-Gated Ion Channels
brain sciences Review Interplay between Gating and Block of Ligand-Gated Ion Channels Matthew B. Phillips 1,2, Aparna Nigam 1 and Jon W. Johnson 1,2,* 1 Department of Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260, USA; [email protected] (M.B.P.); [email protected] (A.N.) 2 Center for Neuroscience, University of Pittsburgh, Pittsburgh, PA 15260, USA * Correspondence: [email protected]; Tel.: +1-(412)-624-4295 Received: 27 October 2020; Accepted: 26 November 2020; Published: 1 December 2020 Abstract: Drugs that inhibit ion channel function by binding in the channel and preventing current flow, known as channel blockers, can be used as powerful tools for analysis of channel properties. Channel blockers are used to probe both the sophisticated structure and basic biophysical properties of ion channels. Gating, the mechanism that controls the opening and closing of ion channels, can be profoundly influenced by channel blocking drugs. Channel block and gating are reciprocally connected; gating controls access of channel blockers to their binding sites, and channel-blocking drugs can have profound and diverse effects on the rates of gating transitions and on the stability of channel open and closed states. This review synthesizes knowledge of the inherent intertwining of block and gating of excitatory ligand-gated ion channels, with a focus on the utility of channel blockers as analytic probes of ionotropic glutamate receptor channel function. Keywords: ligand-gated ion channel; channel block; channel gating; nicotinic acetylcholine receptor; ionotropic glutamate receptor; AMPA receptor; kainate receptor; NMDA receptor 1. Introduction Neuronal information processing depends on the distribution and properties of the ion channels found in neuronal membranes. -

Molecular Biology of Neuronal Voltage-Gated Calcium Channels
EXPERIMENTAL and MOLECULAR MEDICINE, Vol. 30, No 3, 123-130, September 1998 Molecular biology of neuronal voltage-gated calcium channels Hemin Chin and is capable of directing expression of calcium channel activity in heterologous expression systems. In the central Genetics Research Branch, Division of Basic and Clinical Neuroscience Research, nervous system (CNS), VGCCs are expressed by five National Institute of Mental Health, National Institutes of Health, Bethesda, Maryland, distinct a1 subunit genes (α1A, α1B, α1C, α1D and α1E), U.S.A. which exhibit further variations due to alternative splicing of the primary RNA transcripts. The α1C and, α1D su b u n i t Accepted 3 August 1998 genes encode dihydropyridine (DHP)-sensitive L-type channels, while the three other α1 subunit genes (α1A, α1B and α1E) give rise to DHP-insensitive P/Q-, N- and R-type channels, respectively. The α2 and δ s u b u n i t proteins are produced by proteolytic cleavage of a larger precursor produced by the single α2-δ gene (Table 1). Introduction Three alternatively spliced variants of the α2 subunit are expressed in a tissue-specific manner. Two variants Calcium ions are important intracellular messengers have been isolated from the brain and skeletal muscle mediating a number of neuronal functions including neuro- (Kim et al., 1992; Williams et al., 1992), and a distinct transmitter release, neurosecretion, neuronal excitation, third splice variant which is expressed in glial cells has survival of eurons, and regulation of gene expression. been recently identified (Puro et al., 1996). In addition to The entry of calcium across the plasmamembrane in the gene encoding the skeletal muscle β subunit, three response to membrane depolarization or activation of 1 other β subunit genes (β2, β3 and β4) have been isolated neurotransmitter receptors represents a major pathway thus far. -

Expression Profiling of Ion Channel Genes Predicts Clinical Outcome in Breast Cancer
UCSF UC San Francisco Previously Published Works Title Expression profiling of ion channel genes predicts clinical outcome in breast cancer Permalink https://escholarship.org/uc/item/1zq9j4nw Journal Molecular Cancer, 12(1) ISSN 1476-4598 Authors Ko, Jae-Hong Ko, Eun A Gu, Wanjun et al. Publication Date 2013-09-22 DOI http://dx.doi.org/10.1186/1476-4598-12-106 Peer reviewed eScholarship.org Powered by the California Digital Library University of California Ko et al. Molecular Cancer 2013, 12:106 http://www.molecular-cancer.com/content/12/1/106 RESEARCH Open Access Expression profiling of ion channel genes predicts clinical outcome in breast cancer Jae-Hong Ko1, Eun A Ko2, Wanjun Gu3, Inja Lim1, Hyoweon Bang1* and Tong Zhou4,5* Abstract Background: Ion channels play a critical role in a wide variety of biological processes, including the development of human cancer. However, the overall impact of ion channels on tumorigenicity in breast cancer remains controversial. Methods: We conduct microarray meta-analysis on 280 ion channel genes. We identify candidate ion channels that are implicated in breast cancer based on gene expression profiling. We test the relationship between the expression of ion channel genes and p53 mutation status, ER status, and histological tumor grade in the discovery cohort. A molecular signature consisting of ion channel genes (IC30) is identified by Spearman’s rank correlation test conducted between tumor grade and gene expression. A risk scoring system is developed based on IC30. We test the prognostic power of IC30 in the discovery and seven validation cohorts by both Cox proportional hazard regression and log-rank test. -

Co-Assembly of N-Type Ca and BK Channels Underlies Functional
Research Article 985 Co-assembly of N-type Ca2+ and BK channels underlies functional coupling in rat brain David J. Loane*, Pedro A. Lima‡ and Neil V. Marrion§ Department of Pharmacology and MRC Centre for Synaptic Plasticity, University of Bristol, Bristol, BS8 1TD, UK *Present address: Laboratory for the Study of CNS Injury, Department of Neuroscience, Georgetown University Medical Center, Washington, DC 20057, USA ‡Present address: Dep. Fisiologia, Fac. Ciências Médicas, UNL, 1169-056 Lisboa, Portugal §Author for correspondence (e-mail: [email protected]) Accepted 9 January 2007 Journal of Cell Science 120, 985-995 Published by The Company of Biologists 2007 doi:10.1242/jcs.03399 Summary Activation of large conductance Ca2+-activated potassium and reproduced the interaction. Co-expression of (BK) channels hastens action potential repolarisation and CaV2.2/CaV3 subunits with Slo27 channels revealed rapid generates the fast afterhyperpolarisation in hippocampal functional coupling. By contrast, extremely rare examples pyramidal neurons. A rapid coupling of Ca2+ entry with of rapid functional coupling were observed with co- BK channel activation is necessary for this to occur, which expression of CaV1.2/CaV3 and Slo27 channels. Action might result from an identified coupling of Ca2+ entry potential repolarisation in hippocampal pyramidal neurons through N-type Ca2+ channels to BK channel activation. was slowed by the N-type channel blocker -conotoxin This selective coupling was extremely rapid and resistant GVIA, but not by the L-type channel blocker isradipine. to intracellular BAPTA, suggesting that the two channel These data showed that selective functional coupling types are close.