Cyb5r3 Links Foxo1-Dependent Mitochondrial Dysfunction with Β-Cell Failure
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CYB5R3 Gene Cytochrome B5 Reductase 3
CYB5R3 gene cytochrome b5 reductase 3 Normal Function The CYB5R3 gene provides instruction for making an enzyme called cytochrome b5 reductase 3. This enzyme is involved in transferring negatively charged particles called electrons from one molecule to another. Two versions (isoforms) of this enzyme are produced from the CYB5R3 gene. The soluble isoform is present only in red blood cells, and the membrane-bound isoform is found in all other cell types. Normal red blood cells contain molecules of iron-containing hemoglobin, which deliver oxygen to the body's tissues. The iron in hemoglobin is ferrous (Fe2+), but it can spontaneously become ferric (Fe3+). Hemoglobin that contains ferric iron is called methemoglobin, and it cannot deliver oxygen. The soluble isoform of cytochrome b5 reductase 3 changes ferric iron back to ferrous iron so hemoglobin can function. Normally, red blood cells contain less than 2 percent methemoglobin. The membrane-bound isoform is embedded in the membranes of various cellular compartments and is widely used in the body. This isoform is necessary for many chemical reactions, including the breakdown and formation of fatty acids, the formation of cholesterol, and the breakdown of various molecules and drugs. Health Conditions Related to Genetic Changes Autosomal recessive congenital methemoglobinemia More than 65 mutations in the CYB5R3 gene have been found to cause autosomal recessive congenital methemoglobinemia types I and II. Most of these CYB5R3 gene mutations cause autosomal recessive congenital methemoglobinemia type I, which is characterized by a lack of oxygen in the body's tissues and bluish appearance of the skin, lips, and nails (cyanosis). -
Supplementary Tables and Figures
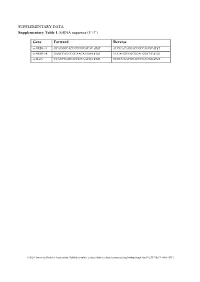
SUPPLEMENTARY DATA Supplementary Table 1. SiRNA sequence (5’-3’) Gene Forward Reverse si-HRD1-1# GCAUGGCAGUCCUGUACAU dTdT AUGUACAGGACUGCCAUGC dTdT si-HRD1-2# GAGCCAUCCGCAACAUGAA dTdT UUCAUGUUGCGGAUGGCUC dTdT si-MafA CCAUCGAGUACGUCAACGA dTdT UCGUUGACGUACUCGAUGG dTdT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 2. Primer sequences for qRT-PCR (5’-3’) Gene Forward Reverse human HRD1 GCTCACGCCTACTACCTCAAA GCCAGACAAGTCTCTGTGACG mouse mafA AAGCGGCGCACGCTCAAGAA GGTCCCGCTCCTTGGCCAGA mouse insulin1 CACTTCCTACCCCTGCTGG ACCACAAAGATGCTGTTTGACA mouse β-actin AGGCCAACCGTGAAAAGATG AGAGCATAGCCCTCGTAGATGG human β-actin CATGTACGTTGCTATCCAGGC CTCCTTAATGTCACGCACGAT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 3. Primer sequences for ChIP (5’-3’) Gene promoter Forward Reverse mouse Insulin1, 2 GGAACTGTGAAACAGTCCAAGG CCCCCTGGACTTTGCTGTTTG ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 4. Primer sequences for PCR (5’-3’) Gene Forward Reverse HRD1-pDsred CCCAAGCTTATGTTCCGCACCGCAGT GGGGTACCCAGTGGGCAACAGGGG HRD1-pCMV- Flag GGGGTACCATGTTCCGCACCGCAGT CCCAAGCTTGTGGGCAACAGGGGACT C HRD1-pCMV-HA GGCCATGGGCCATATGGGATCCTTCC AGGGATGCCACCCGGGGATCCTCAGT GCACCGCAGTGATG GGGCAACAGGGGAC HRD1-N-HA GGCCATGGGCCATATGGGATCCTTCC -

Bioinformatics Analysis Based on Gene Expression Omnibus
ANTICANCER RESEARCH 39 : 1689-1698 (2019) doi:10.21873/anticanres.13274 Chemo-resistant Gastric Cancer Associated Gene Expression Signature: Bioinformatics Analysis Based on Gene Expression Omnibus JUN-BAO LIU 1* , TUNYU JIAN 2* , CHAO YUE 3, DAN CHEN 4, WEI CHEN 5, TING-TING BAO 6, HAI-XIA LIU 7, YUN CAO 8, WEI-BING LI 6, ZHIJIAN YANG 9, ROBERT M. HOFFMAN 9 and CHEN YU 6 1Traditional Chinese Medicine Department, People's Hospital of Henan Province, People's Hospital of Zhengzhou University, Zhengzhou, P.R. China; 2Institute of Botany, Jiangsu Province and Chinese Academy of Sciences, Nanjing, P.R. China; 3Department of general surgery, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, P.R. China; 4Research Center of Clinical Oncology, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, P.R. China; 5Department of Head and Neck Surgery, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, P.R. China; 6Department of Integrated TCM & Western Medicine, Jiangsu Cancer Hospital & Jiangsu Institute of Cancer Research & The Affiliated Cancer Hospital of Nanjing Medical University, Nanjing, P.R. China; 7Emergency Department, The Second Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, P.R. China; 8Master candidate of Oncology, Nanjing University of Chinese Medicine, Nanjing, P.R. China; 9AntiCancer, Inc., San Diego, CA, U.S.A. Abstract. Background/Aim: This study aimed to identify identified, including 13 up-regulated and 1,473 down-regulated biomarkers for predicting the prognosis of advanced gastric genes. -

Epigenetic Regulation of Developmental Expression of Cyp2d Genes in Mouse Liver
CORE Metadata, citation and similar papers at core.ac.uk Provided by Elsevier - Publisher Connector Acta Pharmaceutica Sinica B 2012;2(2):146–158 Institute of Materia Medica, Chinese Academy of Medical Sciences Chinese Pharmaceutical Association Acta Pharmaceutica Sinica B www.elsevier.com/locate/apsb www.sciencedirect.com ORIGINAL ARTICLE Epigenetic regulation of developmental expression of Cyp2d genes in mouse liver Ye Lia, Xiao-bo Zhongb,n aDepartment of Pharmacology, School of Chemical Biology and Pharmaceutical Sciences, Capital Medical University, Beijing 100069, China bDepartment of Pharmacology, Toxicology, and Therapeutics, University of Kansas Medical Center, Kansas City, KS 66160, USA Received 28 November 2011; revised 26 December 2011; accepted 10 January 2012 KEY WORDS Abstract CYP2D6 expression in liver is age-dependent. Because epigenetic mechanisms, such as DNA methylation and histone modifications, modulate age-related gene expression during develop- Cyp2d; ment, and are highly conserved among species, the current study examined the epigenetic regulation of DNA methylation; age-related expression of the Cyp2d genes in mouse liver. DNA methylation (DNAme), histone 3 Histone methylation; lysine 4 dimethylation (H3K4me2), and histone 3 lysine 27 trimethylation (H3K27me3) was Liver development established by ChIP-on-chip tiling microarrays from mouse livers at prenatal, neonatal, and adult stages. Levels of DNAme, H3K4me2, and H3K27me3 were analyzed in a genomic region containing the Cyp2d clustering genes and their surrounding genes. Gradually increased expression levels of the Cyp2d9, Cyp2d10, Cyp2d22,andCyp2d26 genes from prenatal, through neonatal, to adult are associated with gradually increased levels of H3K4me2 in the nucleosomes associated with these genes. Gene expression patterns during liver development in several Cyp2d surrounding genes, such as Srebf2, Sept3, Ndufa6, Tcf2, Nfam1,andCyb5r3, could be also explained by changes of DNA methylation, H3K4me2, or H3K27me3 in those genes. -

Structural Basis of Inter-Domain Electron Transfer in Ncb5or, a Redox Enzyme Implicated in Diabetes and Lipid Metabolism
STRUCTURAL BASIS OF INTER-DOMAIN ELECTRON TRANSFER IN NCB5OR, A REDOX ENZYME IMPLICATED IN DIABETES AND LIPID METABOLISM By Bin Deng Submitted to the graduate degree program in Rehabilitation Science and the Graduate Faculty of the University of Kansas in partial fulfillment of the requirements for the degree of Doctor of Philosophy Hao Zhu, Ph.D. (co-chair, advisor) Irina Smirnova, Ph.D. (co-chair) David Benson, Ph.D. (co-advisor) WenFang Wang, Ph.D. Aron Fenton, Ph.D. Date defended: August 17, 2011 The Dissertation Committee for Bin Deng certifies that this is the approved version of the following dissertation STRUCTURAL BASIS OF INTER-DOMAIN ELECTRON TRANSFER IN NCB5OR, A REDOX ENZYME IMPLICATED IN DIABETES AND LIPID METABOLISM Hao Zhu, Ph.D. (co-chair, advisor) Irina Smirnova, Ph.D. (co-chair) Date approved: August 22, 2011 ii ABSTRACT NADH cytochrome b5 oxidoreductase (Ncb5or) is a multi-domain redox enzyme found in all animal tissues and associated with the endoplasmic reticulum (ER). Ncb5or contains (from N-terminus to C terminus) a novel N-terminal region, the b5 domain (Ncb5or-b5), the CS domain, and the b5R domain (Ncb5or-b5R). Ncb5or-b5, the heme binding domain, is homologous to microsomal cytochrome b5 (Cyb5A) and belongs to cytochrome b5 superfamily. Ncb5or-b5R, the FAD (flavin adenine dinucleotide) binding domain, is homologous to cytochrome b5 reductase (Cyb5R3) and belongs to ferredoxin NADP+ reductase superfamily. Both superfamilies are of great biological significance whose members have important functions. The CS domain can be assigned into the heat shock protein 20 (HSP20, or p23) family, whose members are known to mediate protein-protein interactions. -

Identification of C2CD4A As a Human Diabetes Susceptibility Gene with a Role in Β Cell Insulin Secretion
Identification of C2CD4A as a human diabetes susceptibility gene with a role in β cell insulin secretion Taiyi Kuoa, Michael J. Kraakmana, Manashree Damleb,c, Richard Gilla, Mitchell A. Lazarb,c,1, and Domenico Accilia,1 aDepartment of Medicine, Berrie Diabetes Center, Columbia University College of Physicians and Surgeons, New York, NY 10032; bThe Institute for Diabetes, Obesity, and Metabolism, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104; and cDivision of Endocrinology, Diabetes, and Metabolism, Department of Medicine, University of Pennsylvania Perelman School of Medicine, Philadelphia, PA 19104 Contributed by Mitchell A. Lazar, July 31, 2019 (sent for review March 14, 2019; reviewed by Alvin C. Powers and Andrew F. Stewart) Fine mapping and validation of genes causing β cell failure from targets. To circumvent this obstacle, we generated FoxO1- susceptibility loci identified in type 2 diabetes genome-wide asso- GFPVenus (Venus) reporter knockin mice, and utilized 2-photon ciation studies (GWAS) poses a significant challenge. The VPS13C- microscopy to track its subcellular localization in pancreatic β C2CD4A-C2CD4B locus on chromosome 15 confers diabetes suscep- cells. We next performed genome-wide FoxO1 chromatin immu- tibility in every ethnic group studied to date. However, the causative noprecipitation sequencing (ChIP-seq) to identify its genomic gene is unknown. FoxO1 is involved in the pathogenesis of β cell targets as well as superenhancers encompassing FoxO1 sites. A dysfunction, but its link to human diabetes GWAS has not been comparative analysis of human islet and murine β cell super- explored. Here we generated a genome-wide map of FoxO1 super- enhancers revealed C2CD4A, a gene encoding an IL-1β–induced β enhancers in chemically identified cells using 2-photon live-cell nuclear protein (18) embedded among several SNPs conferring imaging to monitor FoxO1 localization. -

Supplementary Materials (PDF)
Proteomics of the mediodorsal thalamic nucleus in gastric ulcer induced by restraint-water-immersion-stress Sheng-Nan Gong, Jian-Ping Zhu, Ying-Jie Ma, Dong-Qin Zhao Table S1. The entire list of 2,853 proteins identified between the control and stressed groups Protein NO Protein name Gene name Accession No LogRatio 1 Tubulin alpha-1A chain Tuba1a TBA1A_RAT 0.2320 2 Spectrin alpha chain, non-erythrocytic 1 Sptan1 A0A0G2JZ69_RAT -0.0291 3 ATP synthase subunit alpha, mitochondrial Atp5f1a ATPA_RAT -0.1155 4 Tubulin beta-2B chain Tubb2b TBB2B_RAT 0.0072 5 Actin, cytoplasmic 2 Actg1 ACTG_RAT 0.0001 Sodium/potassium-transporting ATPase Atp1a2 6 subunit alpha-2 AT1A2_RAT -0.0716 7 Spectrin beta chain Sptbn1 A0A0G2K8W9_RAT -0.1158 8 Clathrin heavy chain 1 Cltc CLH1_RAT 0.0788 9 Dihydropyrimidinase-related protein 2 Dpysl2 DPYL2_RAT -0.0696 10 Glyceraldehyde-3-phosphate dehydrogenase Gapdh G3P_RAT -0.0687 Sodium/potassium-transporting ATPase Atp1a3 11 subunit alpha-3 AT1A3_RAT 0.0391 12 ATP synthase subunit beta, mitochondrial Atp5f1b ATPB_RAT 0.1772 13 Cytoplasmic dynein 1 heavy chain 1 Dync1h1 M0R9X8_RAT 0.0527 14 Myelin basic protein transcript variant N Mbp I7EFB0_RAT 0.0696 15 Microtubule-associated protein Map2 F1LNK0_RAT -0.1053 16 Pyruvate kinase PKM Pkm KPYM_RAT -0.2608 17 D3ZQQ5_RAT 0.0087 18 Plectin Plec F7F9U6_RAT -0.0076 19 14-3-3 protein zeta/delta Ywhaz A0A0G2JV65_RAT -0.2431 20 2',3'-cyclic-nucleotide 3'-phosphodiesterase Cnp CN37_RAT -0.0495 21 Creatine kinase B-type Ckb KCRB_RAT -0.0514 Voltage-dependent anion-selective channel -

Hereditary Methemoglobinemia Nadir Bir Hipoksemi Sebebi; Herediter Methemoglobinemi
JOURNAL OF Journal of Contemporary CONTEMPORARY MEDICINE Medicine DOI: 10.16899/jcm.946473 J Contemp Med 202x;xx(x):xxx-xxx Case Report / Olgu sunumu A Rare Cause of Hypoxemia: Hereditary Methemoglobinemia Nadir bir Hipoksemi Sebebi; Herediter Methemoglobinemi Dilek Ergün1, Ecem Narin Çopur1, Recai Ergün1, Büşra Göksel Tulgar2, Fikret Kanat1 1Selcuk University School of Medicine Department of Chest, Konya, Turkey 2Selcuk University School of Medicine Department of Medical Genetics, Konya, Turkey Abstract Öz Hereditary methemoglobinemia is one of the rare causes of Herediter Methemoglobinemi hpokseminin nadir sebeplerinden hypoxemia. Mutations in the CYB5R3 gene cause autosomal biridir. CYB5R3 geninde oluşan mutasyonlar otozomal resesif recessive hereditary methemoglobinemia. Mostly, symptoms herediter methemoglobinemiye neden olur. Çoğunlukla nefes such as shortness of breath, bruise and and fatique occur. It may darlığı, morarma, halsizlik gibi semptomlar görülebilir. Bazen erişkin not display any symptoms until adult ages. Our case was at the yaşa kadar semptom vermeyebilir. Onsekiz yaşında ellerinde ve age of 18 and had sometimes recurring bruise in hands and lips, dudaklarında zaman zaman tekrarlayan morluk, nefes darlığı, çarpıntısı shortness of breath, palpitations and oxygen saturation (SaO2) was olan olgumuzun, pulseoksimetre ile ölçülen oksijen satürasyonu 85%. Methemoglobin (Methb) level was %20 (N;0-1.5) No cardiac (SpO2) %85 saptandı. Methemoglobin (Methb) düzeyi %20 (N;0-1.5) or pulmonary cause could be detected, which could account for idi. Dispne ve siyanozu açıklayacak kardiyak ve pulmoner neden dyspnea and cyanosis, and due to low saturation and high Methb. saptanmayan olguda satürasyonu düşüklüğü ve methb yüksekliği levels, metheglobinemia was considered and high dose IV ascorbic nedeniyle methemoglobinemi düşünülerek, yüksek doz ıntravenöz acid was administered. -

Overexpression of CYB5R3 and NQO1, Two NAD<Sup>+</Sup>-Producing Enzymes, Mimics Aspects of Caloric Restriction
University of Kentucky UKnowledge Pharmacology and Nutritional Sciences Faculty Pharmacology and Nutritional Sciences Publications 8-2018 Overexpression of CYB5R3 and NQO1, Two NAD+-Producing Enzymes, Mimics Aspects of Caloric Restriction Alberto Diaz-Ruiz National Institute on Aging Michael Lanasa National Institute on Aging Joseph Garcia National Institute on Aging Hector Mora National Institute on Aging Frances Fan National Institute on Aging See next page for additional authors Follow this and additional works at: https://uknowledge.uky.edu/pharmacol_facpub Right click to open a feedback form in a new tab to let us know how this document benefits oy u. Part of the Biochemical Phenomena, Metabolism, and Nutrition Commons, Cell and Developmental Biology Commons, Nutrition Commons, and the Physiology Commons Repository Citation Diaz-Ruiz, Alberto; Lanasa, Michael; Garcia, Joseph; Mora, Hector; Fan, Frances; Martin-Montalvo, Alejandro; Di Francesco, Andrea; Calvo-Rubio, Miguel; Salvador-Pascual, Andrea; Aon, Miguel A.; Fishbein, Kenneth W.; Pearson, Kevin J.; Villalba, Jose Manuel; Navas, Placido; Bernier, Michel; and de Cabo, Rafael, "Overexpression of CYB5R3 and NQO1, Two NAD+-Producing Enzymes, Mimics Aspects of Caloric Restriction" (2018). Pharmacology and Nutritional Sciences Faculty Publications. 71. https://uknowledge.uky.edu/pharmacol_facpub/71 This Article is brought to you for free and open access by the Pharmacology and Nutritional Sciences at UKnowledge. It has been accepted for inclusion in Pharmacology and Nutritional Sciences Faculty Publications by an authorized administrator of UKnowledge. For more information, please contact [email protected]. Authors Alberto Diaz-Ruiz, Michael Lanasa, Joseph Garcia, Hector Mora, Frances Fan, Alejandro Martin-Montalvo, Andrea Di Francesco, Miguel Calvo-Rubio, Andrea Salvador-Pascual, Miguel A. -

LINC00116 Codes for a Mitochondrial Peptide Linking Respiration and Lipid Metabolism
LINC00116 codes for a mitochondrial peptide linking respiration and lipid metabolism Anastasia Chugunovaa,b, Elizaveta Losevaa, Pavel Mazinc,d,e, Aleksandra Mitinac, Tsimafei Navalayeua, Dmitry Bilanf,g, Polina Vishnyakovah, Maria Mareyh, Anna Golovinai, Marina Serebryakovab,i, Philipp Pletneva,b, Maria Rubtsovaa,b,i, Waltraud Mairc, Anna Vanyushkinac, Philipp Khaitovichc, Vsevolod Belousovf,g,j, Mikhail Vysokikhh,i,1, Petr Sergieva,b,i,k,1, and Olga Dontsovaa,b,f,i aDepartment of Chemistry, Lomonosov Moscow State University, 119992 Moscow, Russia; bCenter of Life Sciences, Skolkovo Institute of Science and Technology, Moscow, 143028, Russia; cSkoltech Center for Neurobiology and Brain Restoration, Skolkovo Institute of Science and Technology, Moscow, 143028, Russia; dInstitute for Information Transmission Problems (Kharkevich Institute) RAS, 127051 Moscow, Russia; eFaculty of Computer Science, National Research University Higher School of Economics, 119991 Moscow, Russia; fShemyakin-Ovchinnikov Institute of Bioorganic Chemistry, 117997 Moscow, Russia; gPirogov Russian National Research Medical University, 117997 Moscow, Russia; hResearch Center for Obstetrics, Gynecology and Perinatology, 117198 Moscow, Russia; iBelozersky Institute of Physico-Chemical Biology, Lomonosov Moscow State University, 119992 Moscow, Russia; jInstitute for Cardiovascular Physiology, Georg August University Göttingen, 37073 Göttingen, Germany; and kInstitute of Functional Genomics, Lomonosov Moscow State University, 119992 Moscow, Russia Edited by Igor Ulitsky, Weizmann Institute of Science, and accepted by Editorial Board Member David J. Mangelsdorf January 28, 2019 (received for review June 7, 2018) Genes coding for small peptides have been frequently misanno- proteome is composed of over 1,100 proteins (16), 5% of that tated as long noncoding RNA (lncRNA) genes. Here we have dem- number being small proteins of less than 100 amino acids in onstrated that one such transcript is translated into a 56-amino- length (17). -

Analysis of Key Genes and Pathways Associated with the Pathogenesis of Type 2 Diabetes Mellitus
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.12.456106; this version posted August 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Analysis of key genes and pathways associated with the pathogenesis of Type 2 diabetes mellitus Varun Alur1, Varshita Raju2, Basavaraj Vastrad3, Chanabasayya Vastrad*4, Shivakumar Kotturshetti4 1. Department of Endocrinology, J.J. M Medical College, Davanagere, Karnataka 577004, India. 2. Department of Obstetrics and Gynecology, J.J. M Medical College, Davanagere, Karnataka 577004, India. 3. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 4. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karnataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2021.08.12.456106; this version posted August 17, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Type 2 diabetes mellitus (T2DM) is the most common endocrine disorder which poses a serious threat to human health. This investigation aimed to screen the candidate genes differentially expressed in T2DM by bioinformatics analysis. The expression profiling by high throughput sequencing of GSE81608 dataset was retrieved from the gene expression omnibus (GEO) database and analyzed to identify the differentially expressed genes (DEGs) between T2DM and normal controls. Then, Gene Ontology (GO) and pathway enrichment analysis, protein- protein interaction (PPI) network, modules, miRNA-hub gene regulatory network construction and TF-hub gene regulatory network construction, and topological analysis were performed. -

Proteomic Analysis Identifies Distinct Glomerular Extracellular Matrix In
CLINICAL RESEARCH www.jasn.org Proteomic Analysis Identifies Distinct Glomerular Extracellular Matrix in Collapsing Focal Segmental Glomerulosclerosis Michael L. Merchant,1 Michelle T. Barati,1 Dawn J. Caster ,1 Jessica L. Hata,2 Liliane Hobeika,3 Susan Coventry,2 Michael E. Brier,1 Daniel W. Wilkey,1 Ming Li,1 Ilse M. Rood,4 Jeroen K. Deegens,4 Jack F. Wetzels,4 Christopher P. Larsen,5 Jonathan P. Troost,6 Jeffrey B. Hodgin,7 Laura H. Mariani,8 Matthias Kretzler ,8 Jon B. Klein,1,9 and Kenneth R. McLeish1 Due to the number of contributing authors, the affiliations are listed at the end of this article. ABSTRACT Background The mechanisms leading to extracellular matrix (ECM) replacement of areas of glomerular capillaries in histologic variants of FSGS are unknown. This study used proteomics to test the hypothesis that glomerular ECM composition in collapsing FSGS (cFSGS) differs from that of other variants. Methods ECM proteins in glomeruli from biopsy specimens of patients with FSGS not otherwise specified (FSGS-NOS) or cFSGS and from normal controls were distinguished and quantified using mass spectrom- etry, verified and localized using immunohistochemistry (IHC) and confocal microscopy, and assessed for gene expression. The analysis also quantified urinary excretion of ECM proteins and peptides. Results Of 58 ECM proteins that differed in abundance between cFSGS and FSGS-NOS, 41 were more abundant in cFSGS and 17 in FSGS-NOS. IHC showed that glomerular tuft staining for cathepsin B, ca- thepsin C, and annexin A3 in cFSGS was significantly greater than in other FSGS variants, in minimal change disease, or in membranous nephropathy.