Structural Basis of Inter-Domain Electron Transfer in Ncb5or, a Redox Enzyme Implicated in Diabetes and Lipid Metabolism
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Chapter 5 High-Pressure Stopped-Flow Kinetic Studies of Nnos
Probing the dynamics and conformational landscape of neuronal nitric oxide synthase A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Life Sciences 2013 Anna Sobolewska-Stawiarz Contents Contents ...................................................................................................................... 2 List of Figures ............................................................................................................. 6 List of Tables ............................................................................................................ 10 Abstract ..................................................................................................................... 12 Declaration ................................................................................................................ 13 Copyright Statement ................................................................................................ 13 Acknowledgements ................................................................................................... 14 List of Abbreviations ............................................................................................... 15 List of Amino Acids Abbreviations ........................................................................ 17 CHAPTER 1. INTRODUCTION ........................................................................... 18 1.1 Nitric oxide synthase ......................................................................................... -

1 Accepted Preprint First Posted on 4 July 2018 As Manuscript EJE-18
Page 1 of 26 Accepted Preprint first posted on 4 July 2018 as Manuscript EJE-18-0256 Title page Title: Approaches to molecular genetic diagnosis in the management of differences/disorders of sex development (DSD): position paper of EU COST Action BM 1303 "DSDnet" Authors: L. Audí 1, S.F. Ahmed 2, N. Krone 3, M. Cools 4, K. McElreavey 5, P.M. Holterhus 6, A. Greenfield 7, A. Bashamboo 5, O. Hiort 8, S.A. Wudy 9, R. McGowan 2,10, on behalf of the EU COST Action 1 Growth and Development Research Unit, Vall d’Hebron Research Institute (VHIR), Center for Biomedical Research on Rare Diseases (CIBERER), Instituto de Salud Carlos III, Barcelona, 08035, Spain 2 Developmental Endocrinology Research Group, University of Glasgow, Glasgow, UK 3 Academic Unit of Child Health, Department of Oncology and Metabolism, University of Sheffield, Sheffield Children's Hospital, Western Bank, Sheffield S10 2TH, United Kingdom 4 Department of Paediatric Endocrinology, Ghent University Hospital, Paediatrics and Internal Medicine Research Unit, Ghent University, 9000 Ghent, Belgium 5 Human Developmental Genetics, Institut Pasteur, Paris, France 6 Division of Pediatric Endocrinology and Diabetes, University Hospital of Schleswig-Holstein and Christian Albrechts University, Kiel, Germany 7 Mammalian Genetics Unit, Medical Research Council, Harwell Institute, Oxfordshire, UK 1 Copyright © 2018 European Society of Endocrinology. Page 2 of 26 8 Division of Paediatric Endocrinology and Diabetes. Department of Paediatric and Adolescent Medicine, University of Lübeck, Lübeck, Germany 9 Steroid Research & Mass Spectrometry Unit, Laboratory for Translational Hormone Analytics, Division of Pediatric Endocrinology and Diabetology, Center of Child and Adolescent Medicine, Justus-Liebig-University, Giessen, Germany 10 Department of Clinical Genetics, Laboratories Building, Queen Elizabeth University Hospital, Glasgow, UK Corresponding author: Laura Audí Vall d’Hebron Research Institute (VHIR)- P. -

Assessment of Fatty Acid Desaturase (Fads2) Structure-Function Properties in Fish in the Context of Environmental Adaptations and As a Target for Genetic Engineering
Preprints (www.preprints.org) | NOT PEER-REVIEWED | Posted: 28 January 2020 doi:10.20944/preprints202001.0330.v1 Peer-reviewed version available at Biomolecules 2020, 10, 206; doi:10.3390/biom10020206 Review Assessment of Fatty Acid Desaturase (Fads2) Structure-Function Properties in Fish in the Context of Environmental adaptations and as a Target for Genetic Engineering Zuzana Bláhová 1*, Thomas Nelson Harvey 2, Martin Pšenička 1, Jan Mráz 1 1 University of South Bohemia in České Budějovice, Faculty of Fisheries and Protection of Waters, South Bohemian Research Center of Aquaculture and Biodiversity of Hydrocenoses, Zátiší 728/II, 389 25 Vodňany, Czech Republic 2 Centre for Integrative Genetics, Department of Animal and Aquacultural Sciences, Faculty of Biosciences, Norwegian University of Life Sciences, Å s, Norway * Correspondence: [email protected] Abstract: Fatty acid desaturase 2 (Fads2) is the key enzyme of long chain polyunsaturated fatty acid (LC-PUFA) biosynthesis. Endogenous production of these biomolecules in vertebrates, if present, is insufficient to meet demand. Hence, LC-PUFA are considered as conditionally-essential. At present however, LC-PUFA are globally-limited nutrients due to anthropogenic factors. Attention of research is given therefore to find ways to maximize endogenous LC-PUFA production especially in production species, whereby deeper knowledge on molecular mechanisms of enzymatic steps involved is being generated. This review first briefly informs about the milestones in the history of LC-PUFA essentiality exploration before it focuses on the main aim – to highlight the fascinating Fads2 potential to play roles fundamental to adaptation to novel environmental conditions. Investigations are summarized, which elucidate the evolutionary history of fish Fads2 providing an explanation for the remarkable plasticity of this enzyme in fish. -

CYB5R3 Gene Cytochrome B5 Reductase 3
CYB5R3 gene cytochrome b5 reductase 3 Normal Function The CYB5R3 gene provides instruction for making an enzyme called cytochrome b5 reductase 3. This enzyme is involved in transferring negatively charged particles called electrons from one molecule to another. Two versions (isoforms) of this enzyme are produced from the CYB5R3 gene. The soluble isoform is present only in red blood cells, and the membrane-bound isoform is found in all other cell types. Normal red blood cells contain molecules of iron-containing hemoglobin, which deliver oxygen to the body's tissues. The iron in hemoglobin is ferrous (Fe2+), but it can spontaneously become ferric (Fe3+). Hemoglobin that contains ferric iron is called methemoglobin, and it cannot deliver oxygen. The soluble isoform of cytochrome b5 reductase 3 changes ferric iron back to ferrous iron so hemoglobin can function. Normally, red blood cells contain less than 2 percent methemoglobin. The membrane-bound isoform is embedded in the membranes of various cellular compartments and is widely used in the body. This isoform is necessary for many chemical reactions, including the breakdown and formation of fatty acids, the formation of cholesterol, and the breakdown of various molecules and drugs. Health Conditions Related to Genetic Changes Autosomal recessive congenital methemoglobinemia More than 65 mutations in the CYB5R3 gene have been found to cause autosomal recessive congenital methemoglobinemia types I and II. Most of these CYB5R3 gene mutations cause autosomal recessive congenital methemoglobinemia type I, which is characterized by a lack of oxygen in the body's tissues and bluish appearance of the skin, lips, and nails (cyanosis). -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Download English-US Transcript (PDF)
MITOCW | watch?v=56vQ0S2eAjw SPEAKER 1: The following content is provided under a Creative Commons license. Your support will help MIT OpenCourseWare continue to offer high quality educational resources for free. To make a donation or view additional materials from hundreds of MIT courses, visit MIT OpenCourseWare at ocw.mit.edu. PROFESSOR: Today what I want to do within the lexicon is tell you about nature's most spectacularly beautiful cofactors. And these are formed from vitamin B-12, which you find in your vitamin bottle. OK. So what is the structure of vitamin B-12, and why do I say they are spectacularly beautiful? So it's very hard to see, but if you look at the structure of this, where have you seen a molecule this complicated with five membered rings, each of which has a nitrogen in this? You've seen this when you studied hemoglobin, and you think about heme and proto protoporphyrin IX. If you look at the biosynthetic pathway of heme, a branchpoint of that pathway is to make this ring, which is found in adenosylcobalamin and methylcobalamin, which is what we're going to be focusing on today. And this ring is called the corrin ring. So what I want to do is introduce you a little bit to this corrin ring and what's unusual about it compared to protoporphyrin IX that you've seen before. So the vitamin, as in the case of all vitamins that we've talked about over the course of the semester, is not the actual cofactor used in the enzymatic transformation. -

Mutations in SDHD Lead to Autosomal Recessive Encephalomyopathy And
Downloaded from http://jmg.bmj.com/ on December 5, 2017 - Published by group.bmj.com Genotype-phenotype correlations ORIGINAL ARTICLE Mutations in SDHD lead to autosomal recessive encephalomyopathy and isolated mitochondrial complex II deficiency Christopher Benjamin Jackson,1,2 Jean-Marc Nuoffer,3 Dagmar Hahn,3 Holger Prokisch,4,5 Birgit Haberberger,4 Matthias Gautschi,3,6 Annemarie Häberli,3 Sabina Gallati,1 André Schaller1 ▸ Additional material is ABSTRACT succinate to fumarate, is formed by the two larger published online only. To view Background Defects of the mitochondrial respiratory hydrophilic subunits, SDHA and SDHB, which also please visit the journal online (10.1136/jmedgenet-2013- chain complex II (succinate dehydrogenase (SDH) harbour the redox cofactors that participate in elec- 101932) complex) are extremely rare. Of the four nuclear encoded tron transfer to ubiquinone. The cofactor FAD is 1 proteins composing complex II, only mutations in the covalently bound to SDHA which provides the suc- Division of Human Genetics, fl fi Departments of Paediatrics and 70 kDa avoprotein (SDHA) and the recently identi ed cinate binding site, and SDHB possesses three Fe-S Clinical Research, University of complex II assembly factor (SDHAF1) have been found to centres which mediate the electron transfer to ubi- Bern, Bern, Switzerland be causative for mitochondrial respiratory chain diseases. quinone. The smaller hydrophobic SDHC and 2 Graduate School for Cellular Mutations in the other three subunits (SDHB, SDHC, SDHD subunits constitute the membrane anchor and Biomedical Sciences, SDHD) and the second assembly factor (SDHAF2) have and ubiquinone binding sites of CII and localise University of Bern, Bern, 2–4 Switzerland so far only been associated with hereditary CII to the inner mitochondrial membrane. -

Electron Transport Discovery Four Complexes Complex I: Nadhà Coqh2
BI/CH 422/622 OUTLINE: Pyruvate pyruvate dehydrogenase Krebs’ Cycle How did he figure it out? Overview 8 Steps Citrate Synthase Aconitase Isocitrate dehydrogenase Ketoglutarate dehydrogenase Succinyl-CoA synthetase Succinate dehydrogenase Fumarase Malate dehydrogenase Energetics Regulation Summary Oxidative Phosphorylation Energetics (–0.16 V needed for making ATP) Mitochondria Transport (2.4 kcal/mol needed to transport H+ out) Electron transport Discovery Four Complexes Complex I: NADHà CoQH2 Complex II: Succinateà CoQH2 2+ Complex III: CoQH2à Cytochrome C (Fe ) 2+ Complex IV: Cytochrome C (Fe ) à H2O Electron Transport à O2 Inhibitors of Electron Transport Big Drop! • Inhibitors all stop ET and ATP synthesis: very toxic! Spectral work Big Drop! NADH Cyto-a3 Cyto-c1 Big Drop! Cyto-b Cyto-c Cyto-a Fully reduced Flavin Cyto-c + rotenone + antimycin A 300 350 400 450 500 550 600 650 700 1 Electron Transport Electron-Transport Chain Complexes Contain a Series of Electron Carriers • Better techniques for isolating and handling mitochondria, and isolated various fractions of the inner mitochondrial membrane • Measure E°’ • They corresponded to these large drops, and they contained the redox compounds isolated previously. • When assayed for what reactions they could perform, they could perform certain redox reactions and not others. • When isolated, including isolating the individual redox compounds, and measuring the E°’ for each, it was clear that an electron chain was occurring; like a wire! • Lastly, when certain inhibitors were added, some of the redox reactions could be inhibited and others not. Site of the inhibition could be mapped. Electron Transport Electron-Transport Chain Complexes Contain a Series of Electron Carriers • Better techniques for isolating and handling mitochondria, and isolated various fractions of the inner mitochondrial membrane • Measure E°’ • They corresponded to these large drops, and they contained the redox compounds isolated previously. -
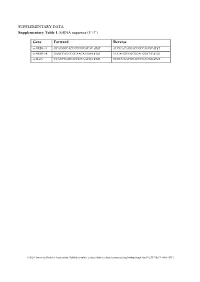
Supplementary Tables and Figures
SUPPLEMENTARY DATA Supplementary Table 1. SiRNA sequence (5’-3’) Gene Forward Reverse si-HRD1-1# GCAUGGCAGUCCUGUACAU dTdT AUGUACAGGACUGCCAUGC dTdT si-HRD1-2# GAGCCAUCCGCAACAUGAA dTdT UUCAUGUUGCGGAUGGCUC dTdT si-MafA CCAUCGAGUACGUCAACGA dTdT UCGUUGACGUACUCGAUGG dTdT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 2. Primer sequences for qRT-PCR (5’-3’) Gene Forward Reverse human HRD1 GCTCACGCCTACTACCTCAAA GCCAGACAAGTCTCTGTGACG mouse mafA AAGCGGCGCACGCTCAAGAA GGTCCCGCTCCTTGGCCAGA mouse insulin1 CACTTCCTACCCCTGCTGG ACCACAAAGATGCTGTTTGACA mouse β-actin AGGCCAACCGTGAAAAGATG AGAGCATAGCCCTCGTAGATGG human β-actin CATGTACGTTGCTATCCAGGC CTCCTTAATGTCACGCACGAT ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 3. Primer sequences for ChIP (5’-3’) Gene promoter Forward Reverse mouse Insulin1, 2 GGAACTGTGAAACAGTCCAAGG CCCCCTGGACTTTGCTGTTTG ©2020 American Diabetes Association. Published online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db19-1060/-/DC1 SUPPLEMENTARY DATA Supplementary Table 4. Primer sequences for PCR (5’-3’) Gene Forward Reverse HRD1-pDsred CCCAAGCTTATGTTCCGCACCGCAGT GGGGTACCCAGTGGGCAACAGGGG HRD1-pCMV- Flag GGGGTACCATGTTCCGCACCGCAGT CCCAAGCTTGTGGGCAACAGGGGACT C HRD1-pCMV-HA GGCCATGGGCCATATGGGATCCTTCC AGGGATGCCACCCGGGGATCCTCAGT GCACCGCAGTGATG GGGCAACAGGGGAC HRD1-N-HA GGCCATGGGCCATATGGGATCCTTCC -
![And Detoxification (Benzola]Pyrene Quinones/Oxygen Radicals/NADPH-Cytochrome P-450 Reductase) PAUL L](https://docslib.b-cdn.net/cover/7447/and-detoxification-benzola-pyrene-quinones-oxygen-radicals-nadph-cytochrome-p-450-reductase-paul-l-837447.webp)
And Detoxification (Benzola]Pyrene Quinones/Oxygen Radicals/NADPH-Cytochrome P-450 Reductase) PAUL L
Proc. Nati. Acad. Sci. USA Vol. 81, pp. 1696-1700, March 1984 Biochemistry Mutagenicity of quinones: Pathways of metabolic activation and detoxification (benzola]pyrene quinones/oxygen radicals/NADPH-cytochrome P-450 reductase) PAUL L. CHESIS*, DAVID E. LEVIN*, MARTYN T. SMITHt, LARS ERNSTERt, AND BRUCE N. AMES* Departments of *Biochemistry and tBiomedical and Environmental Health Sciences, School of Public Health, University of California, Berkeley, CA 94720; and tDepartment of Biochemistry, Arrhenius Laboratory, University of Stockholm, S-10691 Stockholm, Sweden Contributed by Bruce N. Ames, December 12, 1983 ABSTRACT The mutagenicity of various quinones, a class nones might also be mutagenic, and we have tested this pos- of compounds widely distributed in nature, is demonstrated in sibility using the TA104 strain, which is sensitive to a wide the Salmonella TA104 tester strain. The metabolic pathways variety of oxidative mutagens (17). We have also attempted by which four quinones, menadione, benzo[alpyrene 3,6-qui- to characterize the pathways by which several different qui- none, 9,10-phenanthrenequinone, and danthron, caused mu- nones are metabolized and to study the potential mutagenic- tagenicity in this test system were investigated in detail as were ity of the metabolites and side products formed. We there- the detoxification pathways. The two-electron reduction of fore decided to investigate only those quinones that required these quinones by NAD(P)H-quinone oxidoreductase (DT-di- metabolic activation to exhibit mutagenicity. To limit the aphorase) was not mutagenic, whereas the one-electron reduc- scope of this project we also chose not to study quinones tion, catalyzed by NADPH-cytochrome P-450 reductase, was that possess reactive leaving groups. -

Cryoelectron Microscopy Structure and Mechanism of the Membrane-Associated Electron-Bifurcating Flavoprotein Fix/Etfabcx
Cryoelectron microscopy structure and mechanism of the membrane-associated electron-bifurcating flavoprotein Fix/EtfABCX Xiang Fenga, Gerrit J. Schutb, Gina L. Lipscombb, Huilin Lia,1, and Michael W. W. Adamsb,1 aDepartment of Structural Biology, Van Andel Institute, Grand Rapids, MI 49503 and bDepartment of Biochemistry and Molecular Biology, University of Georgia, Athens, GA 30602 Edited by Michael A. Marletta, University of California, Berkeley, CA, and approved November 22, 2020 (received for review August 10, 2020) The electron-transferring flavoprotein-menaquinone oxidoreduc- FBEB enzymes can be divided into four independently tase ABCX (EtfABCX), also known as FixABCX for its role in evolved groups that are unrelated phylogenetically: 1) electron nitrogen-fixing organisms, is a member of a family of electron- transfer flavoproteins containing EtfAB; 2) [FeFe]-hydrogenases transferring flavoproteins that catalyze electron bifurcation. and formate dehydrogenases containing HydABC; 3) hetero- EtfABCX enables endergonic reduction of ferredoxin (E°′ ∼−450 disulfide reductases containing HdrA; and 4) transhydrogenases mV) using NADH (E°′ −320 mV) as the electron donor by coupling containing NfnAB. X-ray–based structures have been reported this reaction to the exergonic reduction of menaquinone (E°′ −80 for three of the four classes: for butyryl-CoA dehydrogenase mV). Here we report the 2.9 Å structure of EtfABCX, a membrane- (EtfAB-Bcd) (5) and caffeyl-CoA reductase [CarCDE; where associated flavin-based electron bifurcation (FBEB) complex, from CarDE corresponds to EtfAB (6)], for NfnAB (7), and for the a thermophilic bacterium. EtfABCX forms a superdimer with two MvhADG–HdrABC complex (8). No structural information is membrane-associated EtfCs at the dimer interface that contain available for a bifurcating HydABC-type enzyme. -

Design and Fine-Tuning Redox Potentials of Metalloproteins Involved in Electron Transfer in Bioenergetics
1 Design and Fine-tuning Redox Potentials of Metalloproteins Involved in Electron Transfer in Bioenergetics Parisa Hosseinzadeh and Yi Lu Abstract: 1. Introduction A significant portion of biological processes are involved with providing vital energy sources such as ATP, and controlling the flow of energy through living systems. These bioenergetics processes, the most important of which are photosynthesis and respiration, require electron transfer (ET) between different redox partners. Metalloproteins are one of the most widely used ET centers in biology. They can be classified into three major classes: cupredoxins, which include type 1 copper (T1Cu) proteins and CuA centers [1- 10], cytochromes [10-17], and iron-sulfur (FeS) proteins [10,18-24]. Each class of ET proteins transfer electrons between different redox partners which possess different reduction potentials (E°) (Figure 1). Therefore, the ET centers need to adjust their E° in a way that matches those of their redox partners. No single class of protein can cover the entire range of physiological E°, which is between ~ -1V, at which protons are reduced to H2, and 1V, at which water is oxidized to O2. Cupredoxins usually function at the high end of the E°, while FeS proteins are mostly involved in ET reactions possessing relatively low E° [10]. The E°’s of FeS proteins overlap significantly with those of cytochromes that often have intermediate E° among the three classes of ET proteins. Figure 1. Reduction potential range of metal centers in electron transfer metalloprotein. Adapted from ref. [10] 2 In this review, we first describe the importance of tuning E° of ET centers, including the metalloproteins described above.