Clinical Chemistry
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mass Spectrometry-Based Proteomics Techniques and Their Application in Ovarian Cancer Research Agata Swiatly, Szymon Plewa, Jan Matysiak and Zenon J
Swiatly et al. Journal of Ovarian Research (2018) 11:88 https://doi.org/10.1186/s13048-018-0460-6 REVIEW Open Access Mass spectrometry-based proteomics techniques and their application in ovarian cancer research Agata Swiatly, Szymon Plewa, Jan Matysiak and Zenon J. Kokot* Abstract Ovarian cancer has emerged as one of the leading cause of gynecological malignancies. So far, the measurement of CA125 and HE4 concentrations in blood and transvaginal ultrasound examination are essential ovarian cancer diagnostic methods. However, their sensitivity and specificity are still not sufficient to detect disease at the early stage. Moreover, applied treatment may appear to be ineffective due to drug-resistance. Because of a high mortality rate of ovarian cancer, there is a pressing need to develop innovative strategies leading to a full understanding of complicated molecular pathways related to cancerogenesis. Recent studies have shown the great potential of clinical proteomics in the characterization of many diseases, including ovarian cancer. Therefore, in this review, we summarized achievements of proteomics in ovarian cancer management. Since the development of mass spectrometry has caused a breakthrough in systems biology, we decided to focus on studies based on this technique. According to PubMed engine, in the years 2008–2010 the number of studies concerning OC proteomics was increasing, and since 2010 it has reached a plateau. Proteomics as a rapidly evolving branch of science may be essential in novel biomarkers discovery, therapy decisions, progression predication, monitoring of drug response or resistance. Despite the fact that proteomics has many to offer, we also discussed some limitations occur in ovarian cancer studies. -
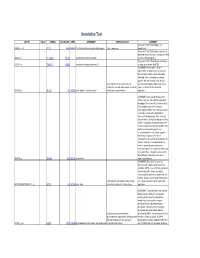
Supplementary Table 5. Functional Annotation of the Largest Gene Cluster(221 Element)
Annotation Tool AFFYID VALUE SYMBOL LOCUSLINK OMIM GENENAME GENEONTOLOGY SUMMARY [Proteome FUNCTION:] Expressed 203054_s_at TCTA 6988 600690 T-cell leukemia translocation altered gene tumor suppressor ubiquitously [Proteome FUNCTION:] May be involved in protein-protein interactions; contains five WD 44563_at FLJ10385 55135 hypothetical protein FLJ10385 domains (WD-40 repeats) [Proteome FUNCTION:] Weakly similarity to 212261_at TNRC15 26058 trinucleotide repeat containing 15 a region of rat nestin (Rn.9701) [SUMMARY:] Actin alpha 1 which is expressed in skeletal muscle is one of six different actin isoforms which have been identified. Actins are highly conserved proteins that are involved in cell motility, actin filament; motor activity; muscle structure and integrity. Alpha actins are a contraction; muscle development; structural major constituent of the contractile 203872_at ACTA1 58 102610 actin, alpha 1, skeletal muscle constituent of cytoskeleton apparatus. [SUMMARY:] Annexin VIII belong to the family of Ca (2+) dependent phospholipid binding proteins (annexins), and has a high 56% identity to annexin V (vascular anticoagulant-alpha). It was initially isolated as 2.2 kb vascular anticoagulant-beta transcript from human placenta, a Ca (2+) dependent phospholipid binding protein that inhibits coagulation and phospholipase A2 activity. However, the fact that annexin VIII is neither an extracellular protein nor associated with the cell surface suggests that it may not play a role in blood coagulation in vivo and its physiological role remains unknown. It is expressed at low levels in human placenta and shows restricted expression in lung endothelia, skin, liver, and kidney. The gene is also found to be selectively overexpressed in acute 203074_at ANXA8 244 602396 annexin A8 myelocytic leukemia. -

1 Supplementary Table S1. Histological And
Supplementary Table S1. Histological and immunocytochemestry of the bcMCF clones. Clones Tumor type AE1 CAM5.2 EMA Vimentin bcMCF-1 Invasive poorly - - - +++ bcMCF-4 differentiated spindle cell type bcMCF-2 Invasive poorly +/- +/- +/- ++ bcMCF-6 differentiated bcMCF-7 epithelial cell type bcMCF-3 Invasive poorly - +/- +/- ++ bcMCF-5 differentiated with mix features of spindle and epithelial type The mouse monoclonal antibodies anti- human cytokeratin of low molecular weight (AE1, Biogenex, San Ramon, CA), cytokeratin peptides 7 and 8 (CAM5.2, Ventana, Tucson, AZ), epithelial membrane antigen (EMA) clone E29 and vimentin, clone V9, both from DakoCytomation Inc. (Fort Collins, CO) were used. Negative (-), weak (+/-), moderate (++) or strong (+++). 1 Supplementary Table S2. Differentially expressed apoptosis genes (GO:00009165) in tumorigenic bcMCF cells. Fold change Symbol Gene name trMCF bcMC caMCF AHR aryl hydrocarbon receptor Ns* -2.1 Ns APP amyloid beta (A4) precursor protein Ns -1.7 -1.7 BAG1 BCL2-associated athanogene Ns -7.6 -9.2 BAG5 BCL2-associated athanogene 5 Ns -1.8 Ns BIRC4 baculoviral IAP repeat-containing 4 Ns -2.0 -2.7 BNIP3L BCL2/adenovirus E1B interacting protein 3-like Ns -2 -1.9 CASP14 caspase 14, apoptosis-related cysteine peptidase Ns -12 -12.3 CASP3 caspase 3, apoptosis-related cysteine peptidase Ns -2.4 Ns CASP6 caspase 6, apoptosis-related cysteine peptidase Ns -4.1 -4.1 CD14 CD14 antigen Ns -4.7 Ns DAPK1 death-associated protein kinase 1 Ns -4.1 -13.4 ELMO3 engulfment and cell motility 3 Ns -9.9 -10.8 ELMOD2 ELMO -

Supporting Information for Proteomics DOI 10.1002/Pmic.200500652
Supporting Information for Proteomics DOI 10.1002/pmic.200500652 Jeffery Forbus, Heidi Spratt, John Wiktorowicz, Zheng Wu, Istvan Boldogh, Larry Denner, Alexander Kurosky, Robert C. Brasier, Bruce Luxon and Allan R. Brasier Functional analysis of the nuclear proteome of human A549 alveolar epithelial cells by HPLC-high resolution 2-D gel electrophoresis ª 2006 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.proteomics-journal.com Supplementary Material Forbus et al., Functional Analysis Of The Nuclear Proteome Of Human A549 Alveolar Epithelial Cells By HPLC- High Resolution 2D Gel Electrophoresis Figure 1. Nuclear Preparation. (A) Microscopic analysis of sucrose step gradient purified nuclei. Purified nuclei were diluted in PBS, plated on a microscope cover slip, and stained with DAPI (Methods). Top panel, high resolution phase contrast microscopy; bottom panel, DAPI staining. Arrows indicate intact nucleoli. (B) Western immunoblot analysis of cytoplasmic and sucrose cushion purified nuclei. Equivalent cell amounts were loaded corresponding to 1 X 106 cells and probed with the indicated antibody, shown at left. Figure 2. Annotated 2-DE template of A549 nuclear proteins. Shown is a master gel representation of the nuclear proteins subjected to HPLC prefractionation and identification by peptide mass fingerprinting. Horizontal dimension, IEF was conducted over a pH range from 3-10. Vertical dimension, fractionation by SDS-PAGE. Migration of molecular weight standards (in kDa) are shown at left. Identity of proteins is shown in Table I. Table I. Annotated proteins in 2-DE template. Shown are high probability identifications from peptide mass fingerprinting using masses measured by MALDI-TOF in a Bayesian algorithm (ProFound). -

Sui Et Al Supplementary Figures
Supplementary Figures Figure S1. Western blot of WI38 fibroblasts treated with fractions of PC3M-LN4 conditioned media eluted from a heparin-sepharose+Cu2+ column with a linear gradient of NaCl plus 20mM imidazole. Supplemental Figure S2. List of proteins present in Tsp-1 repressing fractions (1.0 and 1.1M) and inactive adjacent fractions 0.9M NaCl Keratin 9 1.0M NaCl Keratin 1 Keratin, CK1 1.1M NaCl Keratin, CK2 Keratin 9 Keratin 9 1.2M NaCl Keratin, CK 10 Keratin, CK10 Keratin 1 Keratin 1 Keratin 10 Keratin 2a Keratin, CK10 Keratin 9 Keratin, CK6a Lactotransferrin Keratin, CK 2 Keratin, CK10 Keratin, CK14 precursor Keratin, CK16 57 kDa protein Keratin, CK6e Keratin 1B Keratin, CK6C Keratin 10 Keratin, CK5 Serotransferrin Keratin, CK14 Keratin, CK2 Keratin, CK16 precursor Keratin, CK5 Keratin 1B Cytokeratin type II ALB protein GAPDH Keratin 6L Hornerin similar to KIAA1501 Keratin, CK13 Keratin, type I GAPDH 24- Histone H2A.m cytoskeletal 14 Histone H2A.m dehydrocholesterol Histone H2B.q Keratin 5c Keratin, CK15 reductase precursor Lactotransferrin Keratin, CK3 49 kDa protein Tropomodulin 1 precursor GAPDH Histone H2B.q Protease serine 2 Keratin, Hb4 ALB protein ALB protein isoform B Serotransferrin Hypothetical protein Keratin K6irs Hypothetical protein precursor FLJ20261 Lactotransferrin FLJ90556 Histone 1, H2aa similar to KIAA1501 precursor Splice Isoform 2 of Hypothetical protein similar to KRT8 Histone H4 WD-repeat protein LOC65250 keratin 25 irs1 Serotransferrin 22 DKFZp686J1375 ROK1 precursor Ciliary rootlet Desmoglein-1 Cadherin -

Mass Spectrometric Identification of Ancient Proteins As Potential Molecular Biomarkers for a 2000-Year-Old Osteogenic Sarcoma
Mass Spectrometric Identification of Ancient Proteins as Potential Molecular Biomarkers for a 2000-Year-Old Osteogenic Sarcoma Agnes Bona1, Zoltan Papai1, Gabor Maasz1,2,3,4, Gabor A. Toth5, Eva Jambor1,3, Janos Schmidt1,2,3, Csaba Toth6, Csilla Farkas7, Laszlo Mark1,2,3,4* 1 Department of Analytical Biochemistry, Institute of Biochemistry and Medical Chemistry, University of Pecs, Pecs, Hungary, 2 Janos Szentagothai Research Center, University of Pecs, Pecs, Hungary, 3 Imaging Center for Life and Material Sciences, University of Pecs, Pecs, Hungary, 4 PTE-MTA Human Reproduction Research Group, Pecs, Hungary, 5 Institute of Biology, University of West Hungary, Szombathely, Hungary, 6 Department of Pathology, University Teaching Hospital Markusovszky, Szombathely, Hungary, 7 Department of Archaeology, Vas County Museums’ Directorate, Szombathely, Hungary Abstract Osteosarcoma is the most common primary malignant tumor of bone usually occurring in young adolescent and children. This disease has a poor prognosis, because of the metastases in the period of tumor progression, which are usually developed previous to the clinical diagnosis. In this paper, a 2000-year-old ancient bone remain with osteogenic sarcoma was analyzed searching for tumor biomarkers which are closely related to this disease. After a specific extraction SDS-PAGE gel electrophoresis followed by tryptic digestion was performed. After the digestion the samples were measured using MALDI TOF/TOF MS. Healthy bone samples from same archaeological site were used as control samples. Our results show that in the pathological skeletal remain several well known tumor biomarkers are detected such as annexin A10, BCL-2-like protein, calgizzarin, rho GTPase-activating protein 7, HSP beta-6 protein, transferrin and vimentin compared to the control samples. -

Accession Lit 2DEMS L CMS1 L CMS2 T Otal Accessions T
Supplemental Data Table from "The Human Plasma Proteome: A Non-Redundant List Developed by Combination of Four Separate Sources" by N. Leigh Anderson, Malu Polanski, Rembert Pieper, Tina Gatlin, Radhakrishna S. Tirumalai, Thomas P. Conrads, Timothy D. Veenstra, Joshua N. Adkins, Joel G. Pounds, Richard Fagan, and Anna Lobley (Molecular and Cellular Proteomics, in press (Feb 2004). The table lists all 1175 non-redundant accessions, of which only 195 are detected in more than one source (total_sources > 1). Since a substantial number of the 980 accessions found in only one data set could result from MS identification errors, the reader should view these as candidate plasma components subject to confirmation. CMS1 CMS2 otal_accessions otal_sources Accession Lit 2DEMS L L T T Signal TM Description P29312 0 1 0 0 1 1 no 0 14-3-3 protein zeta/delta (Protein kinase C inhibitor protein- 1) (KCIP-1) (Factor activating exoenzyme S) (FAS). Q9C0C2 0 1 0 0 1 1 no 0 182 kda tankyrase 1-binding protein. P00973 1 0 0 0 1 1 no 0 2'-5'-oligoadenylate synthetase 1 (EC 2.7.7.-) ((2- 5')oligo(A) synthetase 1) (2-5A synthetase 1) (p46/p42 OAS) (E18/E16). P29728 1 0 0 0 1 1 no 0 2'-5'-oligoadenylate synthetase 2 (EC 2.7.7.-) ((2- 5')oligo(A) synthetase 2) (2-5A synthetase 2) (p69 OAS / p71 OAS) (p69oas / p71oas). NP_006178 0 0 0 1 1 1 no 0 2'-5'oligoadenylate synthetase 3; 2'-5'-oligoadenylate synthetase 3 P35998 0 1 0 0 1 1 no 0 26S protease regulatory subunit 7 (MSS1 protein). -

Science Manuscript Template
Supplementary Materials and methods Primary cell cultures Primary human thymic epithelial cell (TEC) cultures were established following the protocol previously described in several published manuscripts (40, 41, 86). Briefly, after removal of the thymic capsule, fresh human thymic tissue was mechanically minced with scissors in Hanks buffer to obtain 0.5 mm explants. Explants were washed twice with hanks buffer to remove as much as possible thymocytes. Explants were set down onto 75 cm2 flasks for 20 min to allow proper adhesion on the flask surface, and grown in RPMI 1640 medium (Invitrogen) supplemented with L-glutamine, Ultroser-g and 20% of horse sera. Minor changes to the published model are provided, the replacement of horse serum by charcoal-treated SVF and the RPMI medium used was a phenol free RPMI medium, in order to avoid phenol hormone-like effects. The percentage of medullary epithelial cells was assessed throughout the culture. Preliminary experiments indicated that the optimal time for high mTEC percentage was between days 6 and 8. As an example of a representative experiment the percentage of mTEC was estimated by immunofluorescence using an anti-Claudin 4 and anti-Keratin 5/14 antibodies, both known as specific markers of mTEC (19, 87, 88) and was over 85% (Supplemental Figure S6). The other cells were either contaminating fibroblasts or residual thymocytes. Thus, all experiments were performed at day 7, in order to achieve experiments only with high enriched mTECs. Under a percentage of 80% cells being Keratin 14 positive, the cell culture was discarded. After 7 days of culture, mTECs were trypsinized and transferred in 24-well plates at 105 cells/well. -

IDENTIFICATION of a NOVEL CANDIDATE RECEPTOR for HUMAN RESPIRATORY SYNCYTL4L VIRUS SUBGROUP a by FARNOOSH TAYYARI M.Sc., The
IDENTIFICATION OF A NOVEL CANDIDATE RECEPTOR FOR HUMAN RESPIRATORY SYNCYTL4L VIRUS SUBGROUP A by FARNOOSH TAYYARI M.Sc., The University of British Columbia, 2003 M.D., Mashhad University of Medical Sciences, Mashhad, Iran, 1997 A THESIS SUBMITTED IN PARTIAL FULFILMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE STUDIES (PATHOLOGY AND LABORATORY MEDICINE) THE UNIVERSITY OF BRITISH COLUMBIA October, 2008 © Farnoosh Tayyari, 2008 Abstract Introduction: Respiratory syncytial virus (RSV) is an important pathogen, especially in children, the elderly, and immunocompromised individuals. Despite RSV being discovered decades ago, there is still no good treatment or prevention for RSV disease. The cell surface receptor for RSV is not known and identification of RSV receptor(s) will provide improved opportunities for understanding the pathogenesis of the viral disease and potential for discovering novel antiviral agents. Hypothesis: RSV infects cells via attachment to cell surface receptor(s) which can be identified by unbiased interrogation of cell membrane constituents and functionally characterized by blocking and competition experiments. Specific Aims: Chemical characterization of RSV receptor(s) by cell surface enzyme treatments, identifying candidate receptor(s), and confirming that any identified candidate has characteristics of a receptor were specific aims of the project. Methods: Chemical characteristics of RSV binding molecule(s) were investigated using enzyme digestion studies. Methods used for identification of candidate receptors included: co immunoprecipitation of candidate RSV receptors using whole virion; purification of RSV surface proteins (either by chromatography or by cloning), and virus overlay protein binding assay (VOPBA) combined with mass spectrometry (MS) and protein database searching. -

Program in Human Neutrophils Fails To
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 is online at: average * The Journal of Immunology Anaplasma phagocytophilum , 20 of which you can access for free at: 2005; 174:6364-6372; ; from submission to initial decision 4 weeks from acceptance to publication J Immunol doi: 10.4049/jimmunol.174.10.6364 http://www.jimmunol.org/content/174/10/6364 Insights into Pathogen Immune Evasion Mechanisms: Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils Dori L. Borjesson, Scott D. Kobayashi, Adeline R. Whitney, Jovanka M. Voyich, Cynthia M. Argue and Frank R. DeLeo cites 28 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2005/05/03/174.10.6364.DC1 This article http://www.jimmunol.org/content/174/10/6364.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* • Why • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2005 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Insights into Pathogen Immune Evasion Mechanisms: Anaplasma phagocytophilum Fails to Induce an Apoptosis Differentiation Program in Human Neutrophils1 Dori L. -

Mass Spectrometry-Based Proteomics Analysis of Bioactive Proteins in EMD That Modulate Adhesion of Gingival Fibroblast to Improve Bio-Integration of Dental Implants
Western University Scholarship@Western Electronic Thesis and Dissertation Repository 3-28-2019 3:00 PM Mass Spectrometry-Based Proteomics Analysis Of Bioactive Proteins In EMD That Modulate Adhesion Of Gingival Fibroblast To Improve Bio-Integration Of Dental Implants David Zuanazzi Machado Jr The University of Western Ontario Supervisor Siqueira, Walter L. The University of Western Ontario Graduate Program in Biochemistry A thesis submitted in partial fulfillment of the equirr ements for the degree in Doctor of Philosophy © David Zuanazzi Machado Jr 2019 Follow this and additional works at: https://ir.lib.uwo.ca/etd Part of the Biochemistry Commons, Dental Materials Commons, Oral Biology and Oral Pathology Commons, and the Periodontics and Periodontology Commons Recommended Citation Zuanazzi Machado, David Jr, "Mass Spectrometry-Based Proteomics Analysis Of Bioactive Proteins In EMD That Modulate Adhesion Of Gingival Fibroblast To Improve Bio-Integration Of Dental Implants" (2019). Electronic Thesis and Dissertation Repository. 6064. https://ir.lib.uwo.ca/etd/6064 This Dissertation/Thesis is brought to you for free and open access by Scholarship@Western. It has been accepted for inclusion in Electronic Thesis and Dissertation Repository by an authorized administrator of Scholarship@Western. For more information, please contact [email protected]. i ABSTRACT Titanium (Ti) implants are used in dental practice to replace damaged or lost teeth. For effective treatment, the dental implant needs to integrate with the surrounding hard and soft tissues on the implant site. Despite improvements in bone-implant integration that have been achieved through surface modifications, the integration with the soft tissues is still deficient. The oral mucosa that embraces the transmucosal component of the implant only contacts the surface without making a strong attachment with the connective tissue. -

Accessory Publication
ACCESSORY PUBLICATION Supplementary Table 1. Genes up regulated in the skin of both HR and LR animals after tick larval challengee c No. of Signal intensity GenBank elemen t=0 t=24 t=0 t=24 a b d Accession ts (HR) (HR) (LR) (LR) Description CF762806 11 12.05 12.55 11.91 12.86 Bos taurus major allergen BDA20 CF766739 1 13.45 14.28 13.04 13.90 Bos taurus hypothetical protein LOC614848 CF768318 1 7.60 9.28 7.71 8.17 hypothetical protein LOC616768 DW521217 1 7.79 8.96 8.30 8.57 no hit DW521255 1 9.49 10.76 9.67 10.07 no hit DW521231 1 9.05 9.88 9.02 9.81 no hit DW521278 1 14.67 15.23 13.97 15.41 no hit CF118614 1 11.35 12.45 10.51 12.23 no hit CF118821 1 7.89 8.61 7.79 8.39 no hit CF764343 1 9.90 10.27 9.70 10.34 no hit CF762711 6 10.88 11.97 10.98 11.70 TIGR TC262428 CF765160 1 11.41 12.19 10.84 11.72 TIGR TC265994 CF117621 1 12.55 13.90 12.17 13.30 TIGR TC288696 CF769314 4 11.27 11.37 10.02 10.57 alpha-2u globulin PGCL4, transcript variant 1 CF763260 1 14.12 14.45 13.17 14.13 ATP synthase F0 subunit 6 ATP synthase, H+ transporting, mitochondrial F0 complex, CF763884 2 9.20 9.51 8.69 9.38 subunit b, isoform 1 ATPase, H+ transporting, lysosomal 42kDa, V1 subunit C, CF764558 1 11.56 11.97 10.60 11.90 isoform 1 CF766396 1 12.58 13.12 12.01 12.65 B-cell CLL/lymphoma 7C CF763972 1 7.58 7.66 7.43 7.91 CD44 antigen Cold shock domain protein E1 (UNR protein) (N-ras upstream CF117686 1 8.19 9.49 8.28 8.61 gene protein) a In cases where multiple array elements were identified for the same gene, one GenBank Accession ID is selected as representative.