Human Gene Evolution the HUMAN MOLECULAR GENETICS Series
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Mass Spectrometry-Based Proteomics Techniques and Their Application in Ovarian Cancer Research Agata Swiatly, Szymon Plewa, Jan Matysiak and Zenon J
Swiatly et al. Journal of Ovarian Research (2018) 11:88 https://doi.org/10.1186/s13048-018-0460-6 REVIEW Open Access Mass spectrometry-based proteomics techniques and their application in ovarian cancer research Agata Swiatly, Szymon Plewa, Jan Matysiak and Zenon J. Kokot* Abstract Ovarian cancer has emerged as one of the leading cause of gynecological malignancies. So far, the measurement of CA125 and HE4 concentrations in blood and transvaginal ultrasound examination are essential ovarian cancer diagnostic methods. However, their sensitivity and specificity are still not sufficient to detect disease at the early stage. Moreover, applied treatment may appear to be ineffective due to drug-resistance. Because of a high mortality rate of ovarian cancer, there is a pressing need to develop innovative strategies leading to a full understanding of complicated molecular pathways related to cancerogenesis. Recent studies have shown the great potential of clinical proteomics in the characterization of many diseases, including ovarian cancer. Therefore, in this review, we summarized achievements of proteomics in ovarian cancer management. Since the development of mass spectrometry has caused a breakthrough in systems biology, we decided to focus on studies based on this technique. According to PubMed engine, in the years 2008–2010 the number of studies concerning OC proteomics was increasing, and since 2010 it has reached a plateau. Proteomics as a rapidly evolving branch of science may be essential in novel biomarkers discovery, therapy decisions, progression predication, monitoring of drug response or resistance. Despite the fact that proteomics has many to offer, we also discussed some limitations occur in ovarian cancer studies. -

Studies on the Proteome of Human Hair - Identifcation of Histones and Deamidated Keratins Received: 15 August 2017 Sunil S
www.nature.com/scientificreports OPEN Studies on the Proteome of Human Hair - Identifcation of Histones and Deamidated Keratins Received: 15 August 2017 Sunil S. Adav 1, Roopa S. Subbaiaih2, Swat Kim Kerk 2, Amelia Yilin Lee 2,3, Hui Ying Lai3,4, Accepted: 12 January 2018 Kee Woei Ng3,4,7, Siu Kwan Sze 1 & Artur Schmidtchen2,5,6 Published: xx xx xxxx Human hair is laminar-fbrous tissue and an evolutionarily old keratinization product of follicle trichocytes. Studies on the hair proteome can give new insights into hair function and lead to the development of novel biomarkers for hair in health and disease. Human hair proteins were extracted by detergent and detergent-free techniques. We adopted a shotgun proteomics approach, which demonstrated a large extractability and variety of hair proteins after detergent extraction. We found an enrichment of keratin, keratin-associated proteins (KAPs), and intermediate flament proteins, which were part of protein networks associated with response to stress, innate immunity, epidermis development, and the hair cycle. Our analysis also revealed a signifcant deamidation of keratin type I and II, and KAPs. The hair shafts were found to contain several types of histones, which are well known to exert antimicrobial activity. Analysis of the hair proteome, particularly its composition, protein abundances, deamidated hair proteins, and modifcation sites, may ofer a novel approach to explore potential biomarkers of hair health quality, hair diseases, and aging. Hair is an important and evolutionarily conserved structure. It originates from hair follicles deep within the der- mis and is mainly composed of hair keratins and KAPs, which form a complex network that contributes to the rigidity and mechanical properties. -

Table SD1. Patient Characteristicsa
Table SD1. Patient characteristicsa Patient Sex Age Esophageal Treatment Maximum Cell Maximum Maximum Genotype Food SPT/Ab SPT/F b RAST b Rhinitisc Atopicc Asthmac Alternative diagnosis Dated (year) Disease eosinophils thickness in mast cells lymphocytes anaphylaxis (positive dermatitis /hpf basal layer /hpf /hpf reaction) 1 M 11 NL None 0 3 5 3 Unk No ND ND ND Yes No No Recurrent croup December 2 M 11 NL LTRA 0 3 4 3 Unk No ND ND ND No No Yes Functional abdominal pain May 3 F 9 NL None 0 3 4 3 Unk Unk ND ND ND Unk Unk Unk Functional abdominal pain March 4 M 14 NL None 0 2 6 4 Unk No ND ND ND No No No Vomiting/diarrhea Febuary 5 F 7 NL LTRA 0 3 5 6 Unk Yes 1 3 ND Unk Unk Yes Functional abdominal pain March 6 F 13 NL None 0 2 4 2 Unk No 0 4 ND No No No Functional abdominal pain August 7 M 17 CE PPI 0 4 7 12 Unk Yes 17 4 ND No No Yes None November 8 M 6 CE PPI 0 4 6 10 Unk Unk ND ND ND Unk Unk Unk None June 9 F 16 CE LTRA 3 4 6 8 Unk No ND ND ND No No Yes None January 10 F 13 CE LTRA+PPI 3 5 4 8 Unk No 0 0 ND No No No None August 11 F 11 CE LTRA+PPI 6 4 6 9 Unk Unk ND ND ND Unk Unk Unk None May 12 M 11 EE PPI 24 6 6 12 TT No 2 5 3 Yes No Yes None November 13 F 4 EE PPI 25 6 15 15 TT No 0 0 ND No No No None November 14 M 15 EE None 30 6 24 6 TG Unk 1 1 ND Unk Unk Yes None February 15 M 15 EE None 31 6 15 11 TT No 8 2 ND Yes No Yes None March 16 M 13 EE PPI 32 6 10 25 TG No 0 0 ND No No No None June 17 M 6 EE PPI 40 7 10 21 TT Yes 5 3 0 Unk Unk No None November 18 M 13 EE LTRA 42 7 10 5 TT No 4 5 2 Yes Yes Yes None November 19 F 16 EE LTRA+PPI -
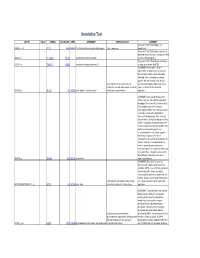
Supplementary Table 5. Functional Annotation of the Largest Gene Cluster(221 Element)
Annotation Tool AFFYID VALUE SYMBOL LOCUSLINK OMIM GENENAME GENEONTOLOGY SUMMARY [Proteome FUNCTION:] Expressed 203054_s_at TCTA 6988 600690 T-cell leukemia translocation altered gene tumor suppressor ubiquitously [Proteome FUNCTION:] May be involved in protein-protein interactions; contains five WD 44563_at FLJ10385 55135 hypothetical protein FLJ10385 domains (WD-40 repeats) [Proteome FUNCTION:] Weakly similarity to 212261_at TNRC15 26058 trinucleotide repeat containing 15 a region of rat nestin (Rn.9701) [SUMMARY:] Actin alpha 1 which is expressed in skeletal muscle is one of six different actin isoforms which have been identified. Actins are highly conserved proteins that are involved in cell motility, actin filament; motor activity; muscle structure and integrity. Alpha actins are a contraction; muscle development; structural major constituent of the contractile 203872_at ACTA1 58 102610 actin, alpha 1, skeletal muscle constituent of cytoskeleton apparatus. [SUMMARY:] Annexin VIII belong to the family of Ca (2+) dependent phospholipid binding proteins (annexins), and has a high 56% identity to annexin V (vascular anticoagulant-alpha). It was initially isolated as 2.2 kb vascular anticoagulant-beta transcript from human placenta, a Ca (2+) dependent phospholipid binding protein that inhibits coagulation and phospholipase A2 activity. However, the fact that annexin VIII is neither an extracellular protein nor associated with the cell surface suggests that it may not play a role in blood coagulation in vivo and its physiological role remains unknown. It is expressed at low levels in human placenta and shows restricted expression in lung endothelia, skin, liver, and kidney. The gene is also found to be selectively overexpressed in acute 203074_at ANXA8 244 602396 annexin A8 myelocytic leukemia. -

AMD) Transmitochondrial Cybrids Protected from Cellular Damage and Death by Human Retinal Progenitor Cells (Hrpcs
Hindawi Stem Cells International Volume 2021, Article ID 6655372, 15 pages https://doi.org/10.1155/2021/6655372 Research Article Age-Related Macular Degeneration (AMD) Transmitochondrial Cybrids Protected from Cellular Damage and Death by Human Retinal Progenitor Cells (hRPCs) Jeffrey J. Yu,1 Daniel B. Azzam ,1 Marilyn Chwa ,1 Kevin Schneider ,1 Jang-Hyeon Cho,1 Chinhui Hsiang,1 Henry Klassen,1 M. Cristina Kenney ,1,2 and Jing Yang 1 1Department of Ophthalmology, Gavin Herbert Eye Institute, University of California Irvine, Irvine, CA 92697, USA 2Department of Pathology and Laboratory Medicine, University of California Irvine, Irvine, CA 92697, USA Correspondence should be addressed to Jing Yang; [email protected] Jeffrey J. Yu and Daniel B. Azzam contributed equally to this work. Received 10 December 2020; Revised 20 January 2021; Accepted 25 January 2021; Published 9 February 2021 Academic Editor: Valeria Sorrenti Copyright © 2021 Jeffrey J. Yu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Purpose. One of the leading causes of irreversible blindness worldwide, age-related macular degeneration (AMD) is a progressive disorder leading to retinal degeneration. While several treatment options exist for the exudative form of AMD, there are currently no FDA-approved treatments for the more common nonexudative (atrophic) form. Mounting evidence suggests that mitochondrial damage and retinal pigment epithelium (RPE) cell death are linked to the pathogenesis of AMD. Human retinal progenitor cells (hRPCs) have been studied as a potential restorative therapy for degenerative conditions of the retina; however, the effects of hRPC treatment on retinal cell survival in AMD have not been elucidated. -

Coding RNA Genes
Review A guide to naming human non-coding RNA genes Ruth L Seal1,2,* , Ling-Ling Chen3, Sam Griffiths-Jones4, Todd M Lowe5, Michael B Mathews6, Dawn O’Reilly7, Andrew J Pierce8, Peter F Stadler9,10,11,12,13, Igor Ulitsky14 , Sandra L Wolin15 & Elspeth A Bruford1,2 Abstract working on non-coding RNA (ncRNA) nomenclature in the mid- 1980s with the approval of initial gene symbols for mitochondrial Research on non-coding RNA (ncRNA) is a rapidly expanding field. transfer RNA (tRNA) genes. Since then, we have worked closely Providing an official gene symbol and name to ncRNA genes brings with experts in the ncRNA field to develop symbols for many dif- order to otherwise potential chaos as it allows unambiguous ferent kinds of ncRNA genes. communication about each gene. The HUGO Gene Nomenclature The number of genes that the HGNC has named per ncRNA class Committee (HGNC, www.genenames.org) is the only group with is shown in Fig 1, and ranges in number from over 4,500 long the authority to approve symbols for human genes. The HGNC ncRNA (lncRNA) genes and over 1,900 microRNA genes, to just four works with specialist advisors for different classes of ncRNA to genes in the vault and Y RNA classes. Every gene symbol has a ensure that ncRNA nomenclature is accurate and informative, Symbol Report on our website, www.genenames.org, which where possible. Here, we review each major class of ncRNA that is displays the gene symbol, gene name, chromosomal location and currently annotated in the human genome and describe how each also includes links to key resources such as Ensembl (Zerbino et al, class is assigned a standardised nomenclature. -

Analysis of Genetic Determinants Associated with Persistent Synthesis of Fetal Hemoglobin
UNIVERSITÀ DEGLI STUDI DI SASSARI PhD School in Biomolecular and Biotechnological Sciences Curriculum: Biochemistry and Molecular Biology Director: Prof. Claudia Crosio “XXVI Ciclo” Analysis of genetic determinants associated with Persistent Synthesis of Fetal Hemoglobin Supervisor: Monica Pirastru, PhD Director: Prof. Claudia Crosio PhD Student: Sandro Trova ................................................................................................................................................. INDEX INDEX ABSTRACT ................................................................................... 3 INTRODUCTION ......................................................................... 4 1. Hemoglobin .......................................................................................... 4 1.1 Structure and function of Hemoglobin ........................................ 4 1.2 Structure of globin genes and their cluster organization ............. 5 1.3 Genomic context of the α– and β–globin gene clusters .............. 9 2. Globin gene switching ....................................................................... 12 2.1 Regulatory regions and transcription factors of globin genes ... 13 2.2 The β–Globin Locus Control Region (β–LCR) role in globin expression ....................................................................... 20 2.3 Chromatin role in β–like globin gene expression: the PYR role .............................................................................. 25 2.4 Summary on the fetal to adult switch ....................................... -

Clinical Chemistry
June 1 2007, Volume 53, Issue 6,pp.999- 1180 Editorials Andrew McCaddon and Peter R. Hudson Methylation and Phosphorylation: A Tangled Relationship? Clin Chem 2007 53: 999-1000. Bob Palais Quantitative Heteroduplex Analysis Clin Chem 2007 53: 1001-1003. Eleftherios P. Diamandis Oncopeptidomics: A Useful Approach for Cancer Diagnosis? Clin Chem 2007 53: 1004-1006. Roger D. Klein The Pain Protective Haplotype: Introducing the Modern Genetic Test Clin Chem 2007 53: 1007-1009. Molecular Diagnostics and Genetics Jörn Lötsch, Inna Belfer, Anja Kirchhof, Bikash K. Mishra, Mitchell B. Max, Alexandra Doehring, Michael Costigan, Clifford J. Woolf, Gerd Geisslinger, and Irmgard Tegeder Reliable Screening for a Pain-Protective Haplotype in the GTP Cyclohydrolase 1 Gene (GCH1) Through the Use of 3 or Fewer Single Nucleotide Polymorphisms Clin Chem 2007 53: 1010-1015. Published online March 15, 2007; 10.1373/clinchem.2006.082883 Kerstin L. Edlefsen, Jonathan F. Tait, Mark H. Wener, and Michael Astion Utilization and Diagnostic Yield of Neurogenetic Testing at a Tertiary Care Facility Clin Chem 2007 53: 1016-1022. Published online April 19, 2007; 10.1373/clinchem.2006.083360 Sara Bremer, Helge Rootwelt, and Stein Bergan Real-Time PCR Determination of IMPDH1 and IMPDH2 Expression in Blood Cells Clin Chem 2007 53: 1023-1029. Published online April 26, 2007; 10.1373/clinchem.2006.081968 Elizabeth Herness Peters, Sandra Rojas-Caro, Mitchell G. Brigell, Robert J. Zahorchak, Shelley Ann des Etages, Patricia L. Ruppel, Charles R. Knight, Bradley Austermiller, Myrna C. Graham, Steve Wowk, Sean Banks, Lakshmi V. Madabusi, Patrick Turk, Donna Wilder, Carole Kempfer, Terry W. Osborn, and James C. -

Hibernation in Pygmy Lorises (Nycticebus Pygmaeus) – What Does It Mean?
Vietnamese Journal of Primatology (2017) vol.2(5), 51-57 Hibernation in pygmy lorises (Nycticebus pygmaeus) – what does it mean? Ulrike Streicher1,3, Julia Nowack2, Gabrielle Stalder2, Christian Walzer2, Tilo Nadler3 and Thomas Ruf2 1 Current address: Cascades Raptor Center, Eugene, USA 2 University of Veterinary Medicine, Research Institute of Wildlife Ecology, Department of Integrative Biology and Evolution, Vienna, Austria, Savoyenstr. 1, 110 Vienna, Austria 3 Endangered Primate Rescue Center, Cuc Phương National Park, Nho Quan District, Ninh Bình Province, Vietnam Corresponding author: Ulrike Streicher <[email protected]> Key words: South-East Asia, primate, torpor, multiday torpor, pygmy loris, hibernation Summary Torpor use in primates appeared to be restricted to African species and was only recently discovered in a species from Asia, the pygmy loris (Nycticebus pygmaeus). This finding has considerable implications for our perception of torpor in this mammal group and demonstrates that torpor is probably more widespread in mammals than commonly thought. This article summarizes the current knowledge on the use of torpor in the pygmy loris and places it into the context of ongoing research on this topic. Hiện tượng ngủ đông ở loài culi nhỏ (Nycticebus pygmaeus) – Ý nghĩa là gì? Tóm tắt Hiện tượng ngủ đông ở các loài linh trưởng được cho rằng chỉ tồn tại ở một số loài linh trưởng ở Châu Phi. Gần đây hiện tượng này được khám phá ở một loài linh trưởng ở Châu Á, loài culi nhỏ (Nycticebus pygmaeus). Phát hiện mới này có thể thay đổi nhận thức của chúng ta về hiện tượng ngủ đông ở nhóm thú này và nó cũng minh chứng rằng hiện tượng ngủ đông có thể phổ biến ở nhiều loài thú khác hơn những gì chúng ta thường nghĩ. -

Discovery of Genes Affecting Resistance of Barley to Adapted And
Douchkov et al. Genome Biology 2014, 15:518 http://genomebiology.com/2014/15/12/518 RESEARCH Open Access Discovery of genes affecting resistance of barley to adapted and non-adapted powdery mildew fungi Dimitar Douchkov1, Stefanie Lück1, Annika Johrde2, Daniela Nowara1, Axel Himmelbach1, Jeyaraman Rajaraman1, Nils Stein1, Rajiv Sharma3, Benjamin Kilian4 and Patrick Schweizer1* Abstract Background: Non-host resistance, NHR, to non-adapted pathogens and quantitative host resistance, QR, confer durable protection to plants and are important for securing yield in a longer perspective. However, a more targeted exploitation of the trait usually possessing a complex mode of inheritance by many quantitative trait loci, QTLs, will require a better understanding of the most important genes and alleles. Results: Here we present results from a transient-induced gene silencing, TIGS, approach of candidate genes for NHR and QR in barley against the powdery mildew fungus Blumeria graminis. Genes were selected based on transcript regulation, multigene-family membership or genetic map position. Out of 1,144 tested RNAi-target genes, 96 significantly affected resistance to the non-adapted wheat- or the compatible barley powdery mildew fungus, with an overlap of four genes. TIGS results for QR were combined with transcript regulation data, allele-trait associations, QTL co-localization and copy number variation resulting in a meta-dataset of 51 strong candidate genes with convergent evidence for a role in QR. Conclusions: This study represents an initial, functional inventory of approximately 3% of the barley transcriptome for a role in NHR or QR against the powdery mildew pathogen. The discovered candidate genes support the idea that QR in this Triticeae host is primarily based on pathogen-associated molecular pattern-triggered immunity, which is compromised by effector molecules produced by the compatible pathogen. -

Gene Network and Pathway Analysis of Transcriptional Signatures Characterizing Sole Ulcer and Digital Dermatitis in Dairy Cows
South Dakota State University Open PRAIRIE: Open Public Research Access Institutional Repository and Information Exchange Electronic Theses and Dissertations 2021 Gene Network and Pathway Analysis of Transcriptional Signatures Characterizing Sole Ulcer and Digital Dermatitis in Dairy Cows Roshin Anie Mohan South Dakota State University Follow this and additional works at: https://openprairie.sdstate.edu/etd Part of the Dairy Science Commons Recommended Citation Mohan, Roshin Anie, "Gene Network and Pathway Analysis of Transcriptional Signatures Characterizing Sole Ulcer and Digital Dermatitis in Dairy Cows" (2021). Electronic Theses and Dissertations. 5278. https://openprairie.sdstate.edu/etd/5278 This Dissertation - Open Access is brought to you for free and open access by Open PRAIRIE: Open Public Research Access Institutional Repository and Information Exchange. It has been accepted for inclusion in Electronic Theses and Dissertations by an authorized administrator of Open PRAIRIE: Open Public Research Access Institutional Repository and Information Exchange. For more information, please contact [email protected]. GENE NETWORK AND PATHWAY ANALYSIS OF TRANSCRIPTIONAL SIGNATURES CHARACTERIZING SOLE ULCER AND DIGITAL DERMATITIS IN DAIRY COWS BY ROSHIN ANIE MOHAN A dissertation submitted in partial fulfillment of the requirements for the Doctor of Philosophy Major in Biological Sciences Specialization in Dairy Science South Dakota State University 2021 ii DISSERTATION ACCEPTANCE PAGE Roshin Anie Mohan This dissertation is approved as a creditable and independent investigation by a candidate for the Doctor of Philosophy degree and is acceptable for meeting the dissertation requirements for this degree. Acceptance of this does not imply that the conclusions reached by the candidate are necessarily the conclusions of the major department. -

Ribosomal RNA
Ribosomal RNA Ribosomal ribonucleic acid (rRNA) is a type of non-coding RNA which is the primary component of ribosomes, essential to all cells. rRNA is a ribozyme which carries out protein synthesis in ribosomes. Ribosomal RNA is transcribed from ribosomal DNA (rDNA) and then bound to ribosomal proteins to form small and large ribosome subunits. rRNA is the physical and mechanical factor of the ribosome that forces transfer RNA (tRNA) and messenger RNA (mRNA) to process and translate the latter into proteins.[1] Ribosomal RNA Three-dimensional views of the ribosome, showing rRNA in dark blue (small subunit) is the predominant form of RNA found in most cells; it makes and dark red (large subunit). Lighter colors up about 80% of cellular RNA despite never being translated represent ribosomal proteins. into proteins itself. Ribosomes are composed of approximately 60% rRNA and 40% ribosomal proteins by mass. Contents Structure Assembly Function Subunits and associated ribosomal RNA In prokaryotes In eukaryotes Biosynthesis In eukaryotes Eukaryotic regulation In prokaryotes Prokaryotic regulation Degradation In eukaryotes In prokaryotes Sequence conservation and stability Significance Human genes See also References External links Structure Although the primary structure of rRNA sequences can vary across organisms, base-pairing within these sequences commonly forms stem-loop configurations. The length and position of these rRNA stem-loops allow them to create three-dimensional rRNA structures that are similar across species.[2] Because of these configurations, rRNA can form tight and specific interactions with ribosomal proteins to form ribosomal subunits. These ribosomal proteins contain basic residues (as opposed to acidic residues) and aromatic residues (i.e.