HOXD3 Enhances Motility and Invasiveness Through the TGF-B-Dependent and -Independent Pathways in A549 Cells
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mass Spectrometry-Based Proteomics Techniques and Their Application in Ovarian Cancer Research Agata Swiatly, Szymon Plewa, Jan Matysiak and Zenon J
Swiatly et al. Journal of Ovarian Research (2018) 11:88 https://doi.org/10.1186/s13048-018-0460-6 REVIEW Open Access Mass spectrometry-based proteomics techniques and their application in ovarian cancer research Agata Swiatly, Szymon Plewa, Jan Matysiak and Zenon J. Kokot* Abstract Ovarian cancer has emerged as one of the leading cause of gynecological malignancies. So far, the measurement of CA125 and HE4 concentrations in blood and transvaginal ultrasound examination are essential ovarian cancer diagnostic methods. However, their sensitivity and specificity are still not sufficient to detect disease at the early stage. Moreover, applied treatment may appear to be ineffective due to drug-resistance. Because of a high mortality rate of ovarian cancer, there is a pressing need to develop innovative strategies leading to a full understanding of complicated molecular pathways related to cancerogenesis. Recent studies have shown the great potential of clinical proteomics in the characterization of many diseases, including ovarian cancer. Therefore, in this review, we summarized achievements of proteomics in ovarian cancer management. Since the development of mass spectrometry has caused a breakthrough in systems biology, we decided to focus on studies based on this technique. According to PubMed engine, in the years 2008–2010 the number of studies concerning OC proteomics was increasing, and since 2010 it has reached a plateau. Proteomics as a rapidly evolving branch of science may be essential in novel biomarkers discovery, therapy decisions, progression predication, monitoring of drug response or resistance. Despite the fact that proteomics has many to offer, we also discussed some limitations occur in ovarian cancer studies. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplemental Materials ZNF281 Enhances Cardiac Reprogramming
Supplemental Materials ZNF281 enhances cardiac reprogramming by modulating cardiac and inflammatory gene expression Huanyu Zhou, Maria Gabriela Morales, Hisayuki Hashimoto, Matthew E. Dickson, Kunhua Song, Wenduo Ye, Min S. Kim, Hanspeter Niederstrasser, Zhaoning Wang, Beibei Chen, Bruce A. Posner, Rhonda Bassel-Duby and Eric N. Olson Supplemental Table 1; related to Figure 1. Supplemental Table 2; related to Figure 1. Supplemental Table 3; related to the “quantitative mRNA measurement” in Materials and Methods section. Supplemental Table 4; related to the “ChIP-seq, gene ontology and pathway analysis” and “RNA-seq” and gene ontology analysis” in Materials and Methods section. Supplemental Figure S1; related to Figure 1. Supplemental Figure S2; related to Figure 2. Supplemental Figure S3; related to Figure 3. Supplemental Figure S4; related to Figure 4. Supplemental Figure S5; related to Figure 6. Supplemental Table S1. Genes included in human retroviral ORF cDNA library. Gene Gene Gene Gene Gene Gene Gene Gene Symbol Symbol Symbol Symbol Symbol Symbol Symbol Symbol AATF BMP8A CEBPE CTNNB1 ESR2 GDF3 HOXA5 IL17D ADIPOQ BRPF1 CEBPG CUX1 ESRRA GDF6 HOXA6 IL17F ADNP BRPF3 CERS1 CX3CL1 ETS1 GIN1 HOXA7 IL18 AEBP1 BUD31 CERS2 CXCL10 ETS2 GLIS3 HOXB1 IL19 AFF4 C17ORF77 CERS4 CXCL11 ETV3 GMEB1 HOXB13 IL1A AHR C1QTNF4 CFL2 CXCL12 ETV7 GPBP1 HOXB5 IL1B AIMP1 C21ORF66 CHIA CXCL13 FAM3B GPER HOXB6 IL1F3 ALS2CR8 CBFA2T2 CIR1 CXCL14 FAM3D GPI HOXB7 IL1F5 ALX1 CBFA2T3 CITED1 CXCL16 FASLG GREM1 HOXB9 IL1F6 ARGFX CBFB CITED2 CXCL3 FBLN1 GREM2 HOXC4 IL1F7 -

Cellular and Molecular Signatures in the Disease Tissue of Early
Cellular and Molecular Signatures in the Disease Tissue of Early Rheumatoid Arthritis Stratify Clinical Response to csDMARD-Therapy and Predict Radiographic Progression Frances Humby1,* Myles Lewis1,* Nandhini Ramamoorthi2, Jason Hackney3, Michael Barnes1, Michele Bombardieri1, Francesca Setiadi2, Stephen Kelly1, Fabiola Bene1, Maria di Cicco1, Sudeh Riahi1, Vidalba Rocher-Ros1, Nora Ng1, Ilias Lazorou1, Rebecca E. Hands1, Desiree van der Heijde4, Robert Landewé5, Annette van der Helm-van Mil4, Alberto Cauli6, Iain B. McInnes7, Christopher D. Buckley8, Ernest Choy9, Peter Taylor10, Michael J. Townsend2 & Costantino Pitzalis1 1Centre for Experimental Medicine and Rheumatology, William Harvey Research Institute, Barts and The London School of Medicine and Dentistry, Queen Mary University of London, Charterhouse Square, London EC1M 6BQ, UK. Departments of 2Biomarker Discovery OMNI, 3Bioinformatics and Computational Biology, Genentech Research and Early Development, South San Francisco, California 94080 USA 4Department of Rheumatology, Leiden University Medical Center, The Netherlands 5Department of Clinical Immunology & Rheumatology, Amsterdam Rheumatology & Immunology Center, Amsterdam, The Netherlands 6Rheumatology Unit, Department of Medical Sciences, Policlinico of the University of Cagliari, Cagliari, Italy 7Institute of Infection, Immunity and Inflammation, University of Glasgow, Glasgow G12 8TA, UK 8Rheumatology Research Group, Institute of Inflammation and Ageing (IIA), University of Birmingham, Birmingham B15 2WB, UK 9Institute of -

Genome-Wide DNA Methylation Analysis of KRAS Mutant Cell Lines Ben Yi Tew1,5, Joel K
www.nature.com/scientificreports OPEN Genome-wide DNA methylation analysis of KRAS mutant cell lines Ben Yi Tew1,5, Joel K. Durand2,5, Kirsten L. Bryant2, Tikvah K. Hayes2, Sen Peng3, Nhan L. Tran4, Gerald C. Gooden1, David N. Buckley1, Channing J. Der2, Albert S. Baldwin2 ✉ & Bodour Salhia1 ✉ Oncogenic RAS mutations are associated with DNA methylation changes that alter gene expression to drive cancer. Recent studies suggest that DNA methylation changes may be stochastic in nature, while other groups propose distinct signaling pathways responsible for aberrant methylation. Better understanding of DNA methylation events associated with oncogenic KRAS expression could enhance therapeutic approaches. Here we analyzed the basal CpG methylation of 11 KRAS-mutant and dependent pancreatic cancer cell lines and observed strikingly similar methylation patterns. KRAS knockdown resulted in unique methylation changes with limited overlap between each cell line. In KRAS-mutant Pa16C pancreatic cancer cells, while KRAS knockdown resulted in over 8,000 diferentially methylated (DM) CpGs, treatment with the ERK1/2-selective inhibitor SCH772984 showed less than 40 DM CpGs, suggesting that ERK is not a broadly active driver of KRAS-associated DNA methylation. KRAS G12V overexpression in an isogenic lung model reveals >50,600 DM CpGs compared to non-transformed controls. In lung and pancreatic cells, gene ontology analyses of DM promoters show an enrichment for genes involved in diferentiation and development. Taken all together, KRAS-mediated DNA methylation are stochastic and independent of canonical downstream efector signaling. These epigenetically altered genes associated with KRAS expression could represent potential therapeutic targets in KRAS-driven cancer. Activating KRAS mutations can be found in nearly 25 percent of all cancers1. -

SUPPLEMENTARY MATERIAL Bone Morphogenetic Protein 4 Promotes
www.intjdevbiol.com doi: 10.1387/ijdb.160040mk SUPPLEMENTARY MATERIAL corresponding to: Bone morphogenetic protein 4 promotes craniofacial neural crest induction from human pluripotent stem cells SUMIYO MIMURA, MIKA SUGA, KAORI OKADA, MASAKI KINEHARA, HIROKI NIKAWA and MIHO K. FURUE* *Address correspondence to: Miho Kusuda Furue. Laboratory of Stem Cell Cultures, National Institutes of Biomedical Innovation, Health and Nutrition, 7-6-8, Saito-Asagi, Ibaraki, Osaka 567-0085, Japan. Tel: 81-72-641-9819. Fax: 81-72-641-9812. E-mail: [email protected] Full text for this paper is available at: http://dx.doi.org/10.1387/ijdb.160040mk TABLE S1 PRIMER LIST FOR QRT-PCR Gene forward reverse AP2α AATTTCTCAACCGACAACATT ATCTGTTTTGTAGCCAGGAGC CDX2 CTGGAGCTGGAGAAGGAGTTTC ATTTTAACCTGCCTCTCAGAGAGC DLX1 AGTTTGCAGTTGCAGGCTTT CCCTGCTTCATCAGCTTCTT FOXD3 CAGCGGTTCGGCGGGAGG TGAGTGAGAGGTTGTGGCGGATG GAPDH CAAAGTTGTCATGGATGACC CCATGGAGAAGGCTGGGG MSX1 GGATCAGACTTCGGAGAGTGAACT GCCTTCCCTTTAACCCTCACA NANOG TGAACCTCAGCTACAAACAG TGGTGGTAGGAAGAGTAAAG OCT4 GACAGGGGGAGGGGAGGAGCTAGG CTTCCCTCCAACCAGTTGCCCCAAA PAX3 TTGCAATGGCCTCTCAC AGGGGAGAGCGCGTAATC PAX6 GTCCATCTTTGCTTGGGAAA TAGCCAGGTTGCGAAGAACT p75 TCATCCCTGTCTATTGCTCCA TGTTCTGCTTGCAGCTGTTC SOX9 AATGGAGCAGCGAAATCAAC CAGAGAGATTTAGCACACTGATC SOX10 GACCAGTACCCGCACCTG CGCTTGTCACTTTCGTTCAG Suppl. Fig. S1. Comparison of the gene expression profiles of the ES cells and the cells induced by NC and NC-B condition. Scatter plots compares the normalized expression of every gene on the array (refer to Table S3). The central line -

Methylome and Transcriptome Maps of Human Visceral and Subcutaneous
www.nature.com/scientificreports OPEN Methylome and transcriptome maps of human visceral and subcutaneous adipocytes reveal Received: 9 April 2019 Accepted: 11 June 2019 key epigenetic diferences at Published: xx xx xxxx developmental genes Stephen T. Bradford1,2,3, Shalima S. Nair1,3, Aaron L. Statham1, Susan J. van Dijk2, Timothy J. Peters 1,3,4, Firoz Anwar 2, Hugh J. French 1, Julius Z. H. von Martels1, Brodie Sutclife2, Madhavi P. Maddugoda1, Michelle Peranec1, Hilal Varinli1,2,5, Rosanna Arnoldy1, Michael Buckley1,4, Jason P. Ross2, Elena Zotenko1,3, Jenny Z. Song1, Clare Stirzaker1,3, Denis C. Bauer2, Wenjia Qu1, Michael M. Swarbrick6, Helen L. Lutgers1,7, Reginald V. Lord8, Katherine Samaras9,10, Peter L. Molloy 2 & Susan J. Clark 1,3 Adipocytes support key metabolic and endocrine functions of adipose tissue. Lipid is stored in two major classes of depots, namely visceral adipose (VA) and subcutaneous adipose (SA) depots. Increased visceral adiposity is associated with adverse health outcomes, whereas the impact of SA tissue is relatively metabolically benign. The precise molecular features associated with the functional diferences between the adipose depots are still not well understood. Here, we characterised transcriptomes and methylomes of isolated adipocytes from matched SA and VA tissues of individuals with normal BMI to identify epigenetic diferences and their contribution to cell type and depot-specifc function. We found that DNA methylomes were notably distinct between diferent adipocyte depots and were associated with diferential gene expression within pathways fundamental to adipocyte function. Most striking diferential methylation was found at transcription factor and developmental genes. Our fndings highlight the importance of developmental origins in the function of diferent fat depots. -

Supplementary File 2A Revised
Supplementary file 2A. Differentially expressed genes in aldosteronomas compared to all other samples, ranked according to statistical significance. Missing values were not allowed in aldosteronomas, but to a maximum of five in the other samples. Acc UGCluster Name Symbol log Fold Change P - Value Adj. P-Value B R99527 Hs.8162 Hypothetical protein MGC39372 MGC39372 2,17 6,3E-09 5,1E-05 10,2 AA398335 Hs.10414 Kelch domain containing 8A KLHDC8A 2,26 1,2E-08 5,1E-05 9,56 AA441933 Hs.519075 Leiomodin 1 (smooth muscle) LMOD1 2,33 1,3E-08 5,1E-05 9,54 AA630120 Hs.78781 Vascular endothelial growth factor B VEGFB 1,24 1,1E-07 2,9E-04 7,59 R07846 Data not found 3,71 1,2E-07 2,9E-04 7,49 W92795 Hs.434386 Hypothetical protein LOC201229 LOC201229 1,55 2,0E-07 4,0E-04 7,03 AA454564 Hs.323396 Family with sequence similarity 54, member B FAM54B 1,25 3,0E-07 5,2E-04 6,65 AA775249 Hs.513633 G protein-coupled receptor 56 GPR56 -1,63 4,3E-07 6,4E-04 6,33 AA012822 Hs.713814 Oxysterol bining protein OSBP 1,35 5,3E-07 7,1E-04 6,14 R45592 Hs.655271 Regulating synaptic membrane exocytosis 2 RIMS2 2,51 5,9E-07 7,1E-04 6,04 AA282936 Hs.240 M-phase phosphoprotein 1 MPHOSPH -1,40 8,1E-07 8,9E-04 5,74 N34945 Hs.234898 Acetyl-Coenzyme A carboxylase beta ACACB 0,87 9,7E-07 9,8E-04 5,58 R07322 Hs.464137 Acyl-Coenzyme A oxidase 1, palmitoyl ACOX1 0,82 1,3E-06 1,2E-03 5,35 R77144 Hs.488835 Transmembrane protein 120A TMEM120A 1,55 1,7E-06 1,4E-03 5,07 H68542 Hs.420009 Transcribed locus 1,07 1,7E-06 1,4E-03 5,06 AA410184 Hs.696454 PBX/knotted 1 homeobox 2 PKNOX2 1,78 2,0E-06 -

140503 IPF Signatures Supplement Withfigs Thorax
Supplementary material for Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis Daryle J. DePianto1*, Sanjay Chandriani1⌘*, Alexander R. Abbas1, Guiquan Jia1, Elsa N. N’Diaye1, Patrick Caplazi1, Steven E. Kauder1, Sabyasachi Biswas1, Satyajit K. Karnik1#, Connie Ha1, Zora Modrusan1, Michael A. Matthay2, Jasleen Kukreja3, Harold R. Collard2, Jackson G. Egen1, Paul J. Wolters2§, and Joseph R. Arron1§ 1Genentech Research and Early Development, South San Francisco, CA 2Department of Medicine, University of California, San Francisco, CA 3Department of Surgery, University of California, San Francisco, CA ⌘Current address: Novartis Institutes for Biomedical Research, Emeryville, CA. #Current address: Gilead Sciences, Foster City, CA. *DJD and SC contributed equally to this manuscript §PJW and JRA co-directed this project Address correspondence to Paul J. Wolters, MD University of California, San Francisco Department of Medicine Box 0111 San Francisco, CA 94143-0111 [email protected] or Joseph R. Arron, MD, PhD Genentech, Inc. MS 231C 1 DNA Way South San Francisco, CA 94080 [email protected] 1 METHODS Human lung tissue samples Tissues were obtained at UCSF from clinical samples from IPF patients at the time of biopsy or lung transplantation. All patients were seen at UCSF and the diagnosis of IPF was established through multidisciplinary review of clinical, radiological, and pathological data according to criteria established by the consensus classification of the American Thoracic Society (ATS) and European Respiratory Society (ERS), Japanese Respiratory Society (JRS), and the Latin American Thoracic Association (ALAT) (ref. 5 in main text). Non-diseased normal lung tissues were procured from lungs not used by the Northern California Transplant Donor Network. -
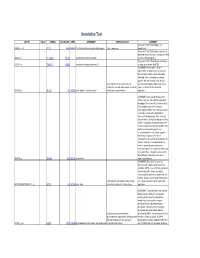
Supplementary Table 5. Functional Annotation of the Largest Gene Cluster(221 Element)
Annotation Tool AFFYID VALUE SYMBOL LOCUSLINK OMIM GENENAME GENEONTOLOGY SUMMARY [Proteome FUNCTION:] Expressed 203054_s_at TCTA 6988 600690 T-cell leukemia translocation altered gene tumor suppressor ubiquitously [Proteome FUNCTION:] May be involved in protein-protein interactions; contains five WD 44563_at FLJ10385 55135 hypothetical protein FLJ10385 domains (WD-40 repeats) [Proteome FUNCTION:] Weakly similarity to 212261_at TNRC15 26058 trinucleotide repeat containing 15 a region of rat nestin (Rn.9701) [SUMMARY:] Actin alpha 1 which is expressed in skeletal muscle is one of six different actin isoforms which have been identified. Actins are highly conserved proteins that are involved in cell motility, actin filament; motor activity; muscle structure and integrity. Alpha actins are a contraction; muscle development; structural major constituent of the contractile 203872_at ACTA1 58 102610 actin, alpha 1, skeletal muscle constituent of cytoskeleton apparatus. [SUMMARY:] Annexin VIII belong to the family of Ca (2+) dependent phospholipid binding proteins (annexins), and has a high 56% identity to annexin V (vascular anticoagulant-alpha). It was initially isolated as 2.2 kb vascular anticoagulant-beta transcript from human placenta, a Ca (2+) dependent phospholipid binding protein that inhibits coagulation and phospholipase A2 activity. However, the fact that annexin VIII is neither an extracellular protein nor associated with the cell surface suggests that it may not play a role in blood coagulation in vivo and its physiological role remains unknown. It is expressed at low levels in human placenta and shows restricted expression in lung endothelia, skin, liver, and kidney. The gene is also found to be selectively overexpressed in acute 203074_at ANXA8 244 602396 annexin A8 myelocytic leukemia. -

1714 Gene Comprehensive Cancer Panel Enriched for Clinically Actionable Genes with Additional Biologically Relevant Genes 400-500X Average Coverage on Tumor
xO GENE PANEL 1714 gene comprehensive cancer panel enriched for clinically actionable genes with additional biologically relevant genes 400-500x average coverage on tumor Genes A-C Genes D-F Genes G-I Genes J-L AATK ATAD2B BTG1 CDH7 CREM DACH1 EPHA1 FES G6PC3 HGF IL18RAP JADE1 LMO1 ABCA1 ATF1 BTG2 CDK1 CRHR1 DACH2 EPHA2 FEV G6PD HIF1A IL1R1 JAK1 LMO2 ABCB1 ATM BTG3 CDK10 CRK DAXX EPHA3 FGF1 GAB1 HIF1AN IL1R2 JAK2 LMO7 ABCB11 ATR BTK CDK11A CRKL DBH EPHA4 FGF10 GAB2 HIST1H1E IL1RAP JAK3 LMTK2 ABCB4 ATRX BTRC CDK11B CRLF2 DCC EPHA5 FGF11 GABPA HIST1H3B IL20RA JARID2 LMTK3 ABCC1 AURKA BUB1 CDK12 CRTC1 DCUN1D1 EPHA6 FGF12 GALNT12 HIST1H4E IL20RB JAZF1 LPHN2 ABCC2 AURKB BUB1B CDK13 CRTC2 DCUN1D2 EPHA7 FGF13 GATA1 HLA-A IL21R JMJD1C LPHN3 ABCG1 AURKC BUB3 CDK14 CRTC3 DDB2 EPHA8 FGF14 GATA2 HLA-B IL22RA1 JMJD4 LPP ABCG2 AXIN1 C11orf30 CDK15 CSF1 DDIT3 EPHB1 FGF16 GATA3 HLF IL22RA2 JMJD6 LRP1B ABI1 AXIN2 CACNA1C CDK16 CSF1R DDR1 EPHB2 FGF17 GATA5 HLTF IL23R JMJD7 LRP5 ABL1 AXL CACNA1S CDK17 CSF2RA DDR2 EPHB3 FGF18 GATA6 HMGA1 IL2RA JMJD8 LRP6 ABL2 B2M CACNB2 CDK18 CSF2RB DDX3X EPHB4 FGF19 GDNF HMGA2 IL2RB JUN LRRK2 ACE BABAM1 CADM2 CDK19 CSF3R DDX5 EPHB6 FGF2 GFI1 HMGCR IL2RG JUNB LSM1 ACSL6 BACH1 CALR CDK2 CSK DDX6 EPOR FGF20 GFI1B HNF1A IL3 JUND LTK ACTA2 BACH2 CAMTA1 CDK20 CSNK1D DEK ERBB2 FGF21 GFRA4 HNF1B IL3RA JUP LYL1 ACTC1 BAG4 CAPRIN2 CDK3 CSNK1E DHFR ERBB3 FGF22 GGCX HNRNPA3 IL4R KAT2A LYN ACVR1 BAI3 CARD10 CDK4 CTCF DHH ERBB4 FGF23 GHR HOXA10 IL5RA KAT2B LZTR1 ACVR1B BAP1 CARD11 CDK5 CTCFL DIAPH1 ERCC1 FGF3 GID4 HOXA11 IL6R KAT5 ACVR2A -
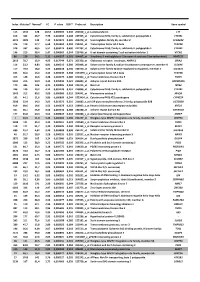
Index Mutated* Normal* FC P Value FDR** Probe Set Description Gene Symbol
Index Mutated* Normal* FC P value FDR** Probe set Description Gene symbol 113 1342 128 10,52 0,000503 0,240 202018_s_at Lactotransferrin LTF 616 362 46,7 7,76 0,003493 0,308 237395_at Cytochrome P450, family 4, subfamily Z, polypeptide 1 CYP4Z1 3073 1009 142 7,10 0,021674 0,385 206378_at Secretoglobin, family 2A, member 2 SCGB2A2 376 119 17,7 6,68 0,001962 0,284 214451_at Transcription factor AP-2 beta TFAP2B 578 337 60,5 5,57 0,003174 0,300 227702_at Cytochrome P450, family 4, subfamily X, polypeptide 1 CYP4X1 146 210 38,4 5,47 0,000669 0,244 219768_at V-set domain containing T cell activation inhibitor 1 VTCN1 86 129 24,2 5,31 0,000327 0,201 204607_at 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 2 (mitochondrial) HMGCS2 2613 78,7 15,9 4,95 0,017744 0,371 205358_at Glutamate receptor, ionotropic, AMPA 2 GRIA2 116 23,2 4,83 4,81 0,000513 0,240 203908_at Solute carrier family 4, sodium bicarbonate cotransporter, member 4 SLC4A4 117 79,4 18,0 4,42 0,000518 0,240 239723_at Solute carrier family 40 (iron-regulated transporter), member 1 SLC40A1 625 56,6 13,0 4,34 0,003549 0,308 1553394_a_atTranscription factor AP-2 beta TFAP2B 413 148 34,6 4,28 0,002175 0,286 240304_s_at Transmembrane channel-like 5 TMC5 5181 216 50,9 4,25 0,039946 0,421 223864_at Ankyrin repeat domain 30A ANKRD30A 179 446 106 4,21 0,000826 0,244 223315_at Netrin 4 NTN4 246 120 29,2 4,12 0,001128 0,251 210096_at Cytochrome P450, family 4, subfamily B, polypeptide 1 CYP4B1 1043 312 80,2 3,89 0,006080 0,319 204041_at Monoamine oxidase B MAOB 192 44,1 11,6 3,80 0,000899 0,244