Reduced X-Linked Diversity in Derived Populations of House Mice
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Mitochondrial Dynamics During Spermatogenesis Grigor Varuzhanyan and David C
© 2020. Published by The Company of Biologists Ltd | Journal of Cell Science (2020) 133, jcs235937. doi:10.1242/jcs.235937 REVIEW SUBJECT COLLECTION: MITOCHONDRIA Mitochondrial dynamics during spermatogenesis Grigor Varuzhanyan and David C. Chan* ABSTRACT Mitophagy is an additional layer of quality control that utilizes Mitochondrial fusion and fission (mitochondrial dynamics) are autophagy (Mizushima, 2007; Mizushima et al., 1998; Tsukada and homeostatic processes that safeguard normal cellular function. This Ohsumi, 1993) to remove excessive mitochondria or damaged relationship is especially strong in tissues with constitutively high energy mitochondria that are beyond repair (Pickles et al., 2018). During demands, such as brain, heart and skeletal muscle. Less is known about mitophagy, microtubule-associated protein 1A/1B light chain 3 the role of mitochondrial dynamics in developmental systems that involve (collectively LC3; also known as MAP1LC3A, MAP1LC3B and changes in metabolic function. One such system is spermatogenesis. MAP1LC3C) is recruited to the autophagosomal membrane and binds The first mitochondrial dynamics gene, Fuzzy onions (Fzo), was to mitochondria that selectively express mitophagy receptors on their discovered in 1997 to mediate mitochondrial fusion during Drosophila outer membrane. This molecular recognition designates damaged spermatogenesis. In mammals, however, the role of mitochondrial fusion mitochondria as cargo for autophagosomes, and the resulting during spermatogenesis remained unknown for nearly two decades after mitophagosomes subsequently fuse with lysosomes for degradation discovery of Fzo. Mammalian spermatogenesis is one of the most and recycling of the engulfed organelles. Much of the molecular complex and lengthy differentiation processes in biology, transforming workings of mitophagy have been parsed in cultured cells, and spermatogonial stem cells into highly specialized sperm cells over a recently developed mouse models are enabling the analysis of 5-week period. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -
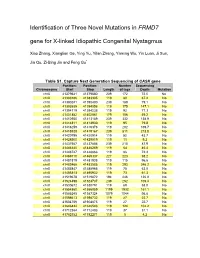
Identification of Three Novel Mutations in FRMD7 Gene for X-Linked Idiopathic Congenital Nystagmus
Identification of Three Novel Mutations in FRMD7 gene for X-linked Idiopathic Congenital Nystagmus Xiao Zhang, Xianglian Ge, Ying Yu, Yilan Zhang, Yaming Wu, Yin Luan, Ji Sun, Jia Qu, Zi-Bing Jin and Feng Gu* Table S1. Capture Next Generation Sequencing of CASK gene Position: Position: Number Sequencing Chromosome Start Stop Length of tags Depth Mutation chrX 41379641 41379880 239 172 72.0 No chrX 41383186 41383305 119 80 67.2 No chrX 41390241 41390480 239 189 79.1 No chrX 41393939 41394058 119 175 147.1 No chrX 41394119 41394238 119 92 77.3 No chrX 41401882 41402061 179 106 59.2 No chrX 41412950 41413189 239 332 138.9 No chrX 41414811 41414930 119 95 79.8 No chrX 41416259 41416378 119 202 169.7 No chrX 41418928 41419167 239 511 213.8 No chrX 41420795 41420914 119 52 43.7 No chrX 41428900 41429019 119 11 9.2 No chrX 41437567 41437806 239 210 87.9 No chrX 41446140 41446259 119 54 45.4 No chrX 41448747 41448866 119 86 72.3 No chrX 41469110 41469337 227 223 98.2 No chrX 41481819 41481938 119 115 96.6 No chrX 41483466 41483585 119 293 246.2 No chrX 41485847 41485966 119 75 63.0 No chrX 41495813 41495932 119 73 61.3 No chrX 41519678 41519872 194 246 126.8 No chrX 41524498 41524737 239 252 105.4 No chrX 41530672 41530791 119 69 58.0 No chrX 41554860 41556059 1199 1932 161.1 No chrX 41586245 41587324 1079 1044 96.8 No chrX 41598613 41598732 119 27 22.7 No chrX 41604756 41604875 119 27 22.7 No chrX 41646424 41646543 119 124 104.2 No chrX 41712364 41712483 119 37 31.1 No chrX 41782152 41782271 119 5 4.2 No Table S2. -

Functional Annotation of the Human Retinal Pigment Epithelium
BMC Genomics BioMed Central Research article Open Access Functional annotation of the human retinal pigment epithelium transcriptome Judith C Booij1, Simone van Soest1, Sigrid MA Swagemakers2,3, Anke HW Essing1, Annemieke JMH Verkerk2, Peter J van der Spek2, Theo GMF Gorgels1 and Arthur AB Bergen*1,4 Address: 1Department of Molecular Ophthalmogenetics, Netherlands Institute for Neuroscience (NIN), an institute of the Royal Netherlands Academy of Arts and Sciences (KNAW), Meibergdreef 47, 1105 BA Amsterdam, the Netherlands (NL), 2Department of Bioinformatics, Erasmus Medical Center, 3015 GE Rotterdam, the Netherlands, 3Department of Genetics, Erasmus Medical Center, 3015 GE Rotterdam, the Netherlands and 4Department of Clinical Genetics, Academic Medical Centre Amsterdam, the Netherlands Email: Judith C Booij - [email protected]; Simone van Soest - [email protected]; Sigrid MA Swagemakers - [email protected]; Anke HW Essing - [email protected]; Annemieke JMH Verkerk - [email protected]; Peter J van der Spek - [email protected]; Theo GMF Gorgels - [email protected]; Arthur AB Bergen* - [email protected] * Corresponding author Published: 20 April 2009 Received: 10 July 2008 Accepted: 20 April 2009 BMC Genomics 2009, 10:164 doi:10.1186/1471-2164-10-164 This article is available from: http://www.biomedcentral.com/1471-2164/10/164 © 2009 Booij et al; licensee BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. -

Genome-Wide Gene Expression Profiling of Randall's Plaques In
CLINICAL RESEARCH www.jasn.org Genome-Wide Gene Expression Profiling of Randall’s Plaques in Calcium Oxalate Stone Formers † † Kazumi Taguchi,* Shuzo Hamamoto,* Atsushi Okada,* Rei Unno,* Hideyuki Kamisawa,* Taku Naiki,* Ryosuke Ando,* Kentaro Mizuno,* Noriyasu Kawai,* Keiichi Tozawa,* Kenjiro Kohri,* and Takahiro Yasui* *Department of Nephro-urology, Nagoya City University Graduate School of Medical Sciences, Nagoya, Japan; and †Department of Urology, Social Medical Corporation Kojunkai Daido Hospital, Daido Clinic, Nagoya, Japan ABSTRACT Randall plaques (RPs) can contribute to the formation of idiopathic calcium oxalate (CaOx) kidney stones; however, genes related to RP formation have not been identified. We previously reported the potential therapeutic role of osteopontin (OPN) and macrophages in CaOx kidney stone formation, discovered using genome-recombined mice and genome-wide analyses. Here, to characterize the genetic patho- genesis of RPs, we used microarrays and immunohistology to compare gene expression among renal papillary RP and non-RP tissues of 23 CaOx stone formers (SFs) (age- and sex-matched) and normal papillary tissue of seven controls. Transmission electron microscopy showed OPN and collagen expression inside and around RPs, respectively. Cluster analysis revealed that the papillary gene expression of CaOx SFs differed significantly from that of controls. Disease and function analysis of gene expression revealed activation of cellular hyperpolarization, reproductive development, and molecular transport in papillary tissue from RPs and non-RP regions of CaOx SFs. Compared with non-RP tissue, RP tissue showed upregulation (˃2-fold) of LCN2, IL11, PTGS1, GPX3,andMMD and downregulation (0.5-fold) of SLC12A1 and NALCN (P,0.01). In network and toxicity analyses, these genes associated with activated mitogen- activated protein kinase, the Akt/phosphatidylinositol 3-kinase pathway, and proinflammatory cytokines that cause renal injury and oxidative stress. -

Table S1. 103 Ferroptosis-Related Genes Retrieved from the Genecards
Table S1. 103 ferroptosis-related genes retrieved from the GeneCards. Gene Symbol Description Category GPX4 Glutathione Peroxidase 4 Protein Coding AIFM2 Apoptosis Inducing Factor Mitochondria Associated 2 Protein Coding TP53 Tumor Protein P53 Protein Coding ACSL4 Acyl-CoA Synthetase Long Chain Family Member 4 Protein Coding SLC7A11 Solute Carrier Family 7 Member 11 Protein Coding VDAC2 Voltage Dependent Anion Channel 2 Protein Coding VDAC3 Voltage Dependent Anion Channel 3 Protein Coding ATG5 Autophagy Related 5 Protein Coding ATG7 Autophagy Related 7 Protein Coding NCOA4 Nuclear Receptor Coactivator 4 Protein Coding HMOX1 Heme Oxygenase 1 Protein Coding SLC3A2 Solute Carrier Family 3 Member 2 Protein Coding ALOX15 Arachidonate 15-Lipoxygenase Protein Coding BECN1 Beclin 1 Protein Coding PRKAA1 Protein Kinase AMP-Activated Catalytic Subunit Alpha 1 Protein Coding SAT1 Spermidine/Spermine N1-Acetyltransferase 1 Protein Coding NF2 Neurofibromin 2 Protein Coding YAP1 Yes1 Associated Transcriptional Regulator Protein Coding FTH1 Ferritin Heavy Chain 1 Protein Coding TF Transferrin Protein Coding TFRC Transferrin Receptor Protein Coding FTL Ferritin Light Chain Protein Coding CYBB Cytochrome B-245 Beta Chain Protein Coding GSS Glutathione Synthetase Protein Coding CP Ceruloplasmin Protein Coding PRNP Prion Protein Protein Coding SLC11A2 Solute Carrier Family 11 Member 2 Protein Coding SLC40A1 Solute Carrier Family 40 Member 1 Protein Coding STEAP3 STEAP3 Metalloreductase Protein Coding ACSL1 Acyl-CoA Synthetase Long Chain Family Member 1 Protein -

Supplemental Table 3 Two-Class Paired Significance Analysis of Microarrays Comparing Gene Expression Between Paired
Supplemental Table 3 Two‐class paired Significance Analysis of Microarrays comparing gene expression between paired pre‐ and post‐transplant kidneys biopsies (N=8). Entrez Fold q‐value Probe Set ID Gene Symbol Unigene Name Score Gene ID Difference (%) Probe sets higher expressed in post‐transplant biopsies in paired analysis (N=1871) 218870_at 55843 ARHGAP15 Rho GTPase activating protein 15 7,01 3,99 0,00 205304_s_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 6,30 4,50 0,00 1563649_at ‐‐ ‐‐ ‐‐ 6,24 3,51 0,00 1567913_at 541466 CT45‐1 cancer/testis antigen CT45‐1 5,90 4,21 0,00 203932_at 3109 HLA‐DMB major histocompatibility complex, class II, DM beta 5,83 3,20 0,00 204606_at 6366 CCL21 chemokine (C‐C motif) ligand 21 5,82 10,42 0,00 205898_at 1524 CX3CR1 chemokine (C‐X3‐C motif) receptor 1 5,74 8,50 0,00 205303_at 3764 KCNJ8 potassium inwardly‐rectifying channel, subfamily J, member 8 5,68 6,87 0,00 226841_at 219972 MPEG1 macrophage expressed gene 1 5,59 3,76 0,00 203923_s_at 1536 CYBB cytochrome b‐245, beta polypeptide (chronic granulomatous disease) 5,58 4,70 0,00 210135_s_at 6474 SHOX2 short stature homeobox 2 5,53 5,58 0,00 1562642_at ‐‐ ‐‐ ‐‐ 5,42 5,03 0,00 242605_at 1634 DCN decorin 5,23 3,92 0,00 228750_at ‐‐ ‐‐ ‐‐ 5,21 7,22 0,00 collagen, type III, alpha 1 (Ehlers‐Danlos syndrome type IV, autosomal 201852_x_at 1281 COL3A1 dominant) 5,10 8,46 0,00 3493///3 IGHA1///IGHA immunoglobulin heavy constant alpha 1///immunoglobulin heavy 217022_s_at 494 2 constant alpha 2 (A2m marker) 5,07 9,53 0,00 1 202311_s_at -

Us 2018 / 0305689 A1
US 20180305689A1 ( 19 ) United States (12 ) Patent Application Publication ( 10) Pub . No. : US 2018 /0305689 A1 Sætrom et al. ( 43 ) Pub . Date: Oct. 25 , 2018 ( 54 ) SARNA COMPOSITIONS AND METHODS OF plication No . 62 /150 , 895 , filed on Apr. 22 , 2015 , USE provisional application No . 62/ 150 ,904 , filed on Apr. 22 , 2015 , provisional application No. 62 / 150 , 908 , (71 ) Applicant: MINA THERAPEUTICS LIMITED , filed on Apr. 22 , 2015 , provisional application No. LONDON (GB ) 62 / 150 , 900 , filed on Apr. 22 , 2015 . (72 ) Inventors : Pål Sætrom , Trondheim (NO ) ; Endre Publication Classification Bakken Stovner , Trondheim (NO ) (51 ) Int . CI. C12N 15 / 113 (2006 .01 ) (21 ) Appl. No. : 15 /568 , 046 (52 ) U . S . CI. (22 ) PCT Filed : Apr. 21 , 2016 CPC .. .. .. C12N 15 / 113 ( 2013 .01 ) ; C12N 2310 / 34 ( 2013. 01 ) ; C12N 2310 /14 (2013 . 01 ) ; C12N ( 86 ) PCT No .: PCT/ GB2016 /051116 2310 / 11 (2013 .01 ) $ 371 ( c ) ( 1 ) , ( 2 ) Date : Oct . 20 , 2017 (57 ) ABSTRACT The invention relates to oligonucleotides , e . g . , saRNAS Related U . S . Application Data useful in upregulating the expression of a target gene and (60 ) Provisional application No . 62 / 150 ,892 , filed on Apr. therapeutic compositions comprising such oligonucleotides . 22 , 2015 , provisional application No . 62 / 150 ,893 , Methods of using the oligonucleotides and the therapeutic filed on Apr. 22 , 2015 , provisional application No . compositions are also provided . 62 / 150 ,897 , filed on Apr. 22 , 2015 , provisional ap Specification includes a Sequence Listing . SARNA sense strand (Fessenger 3 ' SARNA antisense strand (Guide ) Mathew, Si Target antisense RNA transcript, e . g . NAT Target Coding strand Gene Transcription start site ( T55 ) TY{ { ? ? Targeted Target transcript , e . -

Cdna Microarray Analysis and Influx Transporter OATP1B1 in Liver Cells After Exposure to Gadoxetate Disodium, a Gadolinium-Based
in vivo 32 : 677-684 (2018) doi:10.21873/invivo.11293 cDNA Microarray Analysis and Influx Transporter OATP1B1 in Liver Cells After Exposure to Gadoxetate Disodium, a Gadolinium-based Contrast Agent in MRI Liver Imaging CHI-CHENG LU 1* , WEN-KANG CHEN 2* , JO-HUA CHIANG 3, YUH-FENG TSAI 4, YU-NING JUAN 5, PING-CHIN LIN 6, YEU-SHENG TYAN 7,8,9 and JAI-SING YANG 5 1Department of Pharmacy, Buddhist Tzu Chi General Hospital, Hualien, Taiwan, R.O.C.; 2Department of Applied Cosmetology, National Tainan Junior College of Nursing, Tainan, Taiwan, R.O.C.; 3Department of Nursing, Chung Jen Catholic Junior College, Chiayi, Taiwan, R.O.C.; 4Department of Diagnostic Radiology, Shin-Kong Wu Ho-Su Memorial Hospital, Taipei, Taiwan, R.O.C.; 5Department of Medical Research, China Medical University Hospital, China Medical University, Taichung, Taiwan, R.O.C.; 6Department of Medical Imaging, Chia-Yi Christian Hospital, Chiayi, Taiwan, R.O.C.; 7Department of Medical Imaging, Chung Shan Medical University Hospital, Taichung, Taiwan, R.O.C.; 8School of Medical Imaging and Radiological Sciences, 9School of Medicine, Chung Shan Medical University, Taichung, Taiwan, R.O.C. Abstract. Background/Aim: Gadoxetate disodium (Primovist western blotting. Results: Gadoxetate disodium at 5 and 10 or Eovist) is extensively used as a hepatospecific contrast agent mM failed to induce any cell cytotoxicity and morphological during magnetic resonance imaging (MRI) examinations. changes in Chang Liver cells. Our data demonstrated that However, there is no information determining whether gadoxetate disodium significantly enhanced the expression of gadoxetate disodium has a cytotoxic impact and/or affects 29 genes and suppressed that of 27. -

Transcriptional Co-Regulation of Micrornas and Protein-Coding Genes
Transcriptional co-regulation of microRNAs and protein-coding genes A thesis submitted to the University of Manchester for the degree of Doctor of Philosophy in the Faculty of Life Sciences 2013 By Aaron Webber Table of Contents Abstract ......................................................................................................................................... 8 Declaration .................................................................................................................................... 9 Copyright statement ................................................................................................................... 10 Acknowledgements ..................................................................................................................... 11 Preface ........................................................................................................................................ 12 Chapter 1: Introduction .............................................................................................................. 13 1.1 The protein-coding gene expression pathway ............................................................ 13 1.2 Regulation of transcription ......................................................................................... 17 1.2.1 Transcription factors ........................................................................................... 19 1.2.2 Transcriptional regulatory regions ...................................................................... 22 -
Anti-FATE1 Monoclonal Antibody, Clone 8C6 (DCABH-3733) This Product Is for Research Use Only and Is Not Intended for Diagnostic Use
Anti-FATE1 monoclonal antibody, clone 8C6 (DCABH-3733) This product is for research use only and is not intended for diagnostic use. PRODUCT INFORMATION Product Overview Mouse monoclonal to FATE1 Antigen Description The protein encoded by this gene is an enzymatic component of the tricarboxylic acid (TCA) cycle, or Krebs cycle, and catalyzes the formation of L-malate from fumarate. It exists in both a cytosolic form and an N-terminal extended form, differing only in the translation start site used. The N-terminal extended form is targeted to the mitochondrion, where the removal of the extension generates the same form as in the cytoplasm. It is similar to some thermostable class II fumarases and functions as a homotetramer. Mutations in this gene can cause fumarase deficiency and lead to progressive encephalopathy. Immunogen Recombinant full length protein, corresponding to amino acids 1-183 of Human FATE1 produced in HEK293T cells. Isotype IgG1 Source/Host Mouse Species Reactivity Human Clone 8C6 Purity Protein G purified Purification This antibody is purified from Mouse ascites fluid by affinity chromatography. Conjugate Unconjugated Applications WB, IHC-P, ICC/IF Positive Control HEK293T cells transfected with pCMV6-ENTRY FATE1 cDNA; Human ovary adenocarcinoma tissue; COS7 cells transiently transfected by pCMV6-ENTRY FATE1. Format Liquid Size 100 μl Buffer pH: 7.30; Preservative: 0.02% Sodium azide; Constituents: 48% PBS, 50% Glycerol, 1% BSA Preservative 0.02% Sodium Azide 45-1 Ramsey Road, Shirley, NY 11967, USA Email: [email protected] Tel: 1-631-624-4882 Fax: 1-631-938-8221 1 © Creative Diagnostics All Rights Reserved Storage store at -20°C. -
Detection of Circulating Tumor Cells by Reverse Transcription‑Quantitative
5894 MOLECULAR MEDICINE REPORTS 16: 5894-5900, 2017 Detection of circulating tumor cells by reverse transcription‑quantitative polymerase chain reaction and magnetic activated cell sorting in the peripheral blood of patients with hepatocellular carcinoma LIMIN GUO1*, DONGHAI XU2*, YAN LU1, JIRUN PENG3 and LI JIANG1 Departments of 1Hepatobiliary Surgery and 2Radiology, Beijing Ditan Hospital, Capital Medical University, Beijing 100015; 3Department of Surgery, Beijing Shijitan Hospital, Capital Medical University, Beijing 100038, P.R. China Received February 28, 2016; Accepted February 23, 2017 DOI: 10.3892/mmr.2017.7372 Abstract. Hepatocellular carcinoma (HCC) is one of the most sensitivity and specificity in patients with HCC that may be lethal malignancies worldwide. Circulating tumor cells (CTCs) enhanced by MACS. are considered a major cause of recurrence and metastasis in cancer; however, the detection of CTCs is challenging owing Introduction to their very low numbers in peripheral blood (around 10 CTCs per 1,000,000 erythrocytes). Cancer-testis antigens (CTAs) are Hepatocellular carcinoma (HCC) is one of the most lethal specific tumor markers for CTCs. The present study aimed to malignancies worldwide, and is the third most common cause evaluate the sensitivity and specificity of reverse transcrip- of cancer-associated mortality. Previous epidemiological tion-quantitative polymerase chain reaction (RT-qPCR) for the studies revealed that ~626,000 new diagnoses of HCC are detection of nine CTAs as well as placenta‑specific antigen