X-Exome Sequencing of 405 Unresolved Families Identifies Seven
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials
DEPs in osteosarcoma cells comparing to osteoblastic cells Biological Process Protein Percentage of Hits metabolic process (GO:0008152) 29.3 29.3% cellular process (GO:0009987) 20.2 20.2% localization (GO:0051179) 9.4 9.4% biological regulation (GO:0065007) 8 8.0% developmental process (GO:0032502) 7.8 7.8% response to stimulus (GO:0050896) 5.6 5.6% cellular component organization (GO:0071840) 5.6 5.6% multicellular organismal process (GO:0032501) 4.4 4.4% immune system process (GO:0002376) 4.2 4.2% biological adhesion (GO:0022610) 2.7 2.7% apoptotic process (GO:0006915) 1.6 1.6% reproduction (GO:0000003) 0.8 0.8% locomotion (GO:0040011) 0.4 0.4% cell killing (GO:0001906) 0.1 0.1% 100.1% Genes 2179Hits 3870 biological adhesion apoptotic process … reproduction (GO:0000003) , 0.8% (GO:0022610) , 2.7% locomotion (GO:0040011) ,… immune system process cell killing (GO:0001906) , 0.1% (GO:0002376) , 4.2% multicellular organismal process (GO:0032501) , metabolic process 4.4% (GO:0008152) , 29.3% cellular component organization (GO:0071840) , 5.6% response to stimulus (GO:0050896), 5.6% developmental process (GO:0032502) , 7.8% biological regulation (GO:0065007) , 8.0% cellular process (GO:0009987) , 20.2% localization (GO:0051179) , 9. -

Mitochondrial Dynamics During Spermatogenesis Grigor Varuzhanyan and David C
© 2020. Published by The Company of Biologists Ltd | Journal of Cell Science (2020) 133, jcs235937. doi:10.1242/jcs.235937 REVIEW SUBJECT COLLECTION: MITOCHONDRIA Mitochondrial dynamics during spermatogenesis Grigor Varuzhanyan and David C. Chan* ABSTRACT Mitophagy is an additional layer of quality control that utilizes Mitochondrial fusion and fission (mitochondrial dynamics) are autophagy (Mizushima, 2007; Mizushima et al., 1998; Tsukada and homeostatic processes that safeguard normal cellular function. This Ohsumi, 1993) to remove excessive mitochondria or damaged relationship is especially strong in tissues with constitutively high energy mitochondria that are beyond repair (Pickles et al., 2018). During demands, such as brain, heart and skeletal muscle. Less is known about mitophagy, microtubule-associated protein 1A/1B light chain 3 the role of mitochondrial dynamics in developmental systems that involve (collectively LC3; also known as MAP1LC3A, MAP1LC3B and changes in metabolic function. One such system is spermatogenesis. MAP1LC3C) is recruited to the autophagosomal membrane and binds The first mitochondrial dynamics gene, Fuzzy onions (Fzo), was to mitochondria that selectively express mitophagy receptors on their discovered in 1997 to mediate mitochondrial fusion during Drosophila outer membrane. This molecular recognition designates damaged spermatogenesis. In mammals, however, the role of mitochondrial fusion mitochondria as cargo for autophagosomes, and the resulting during spermatogenesis remained unknown for nearly two decades after mitophagosomes subsequently fuse with lysosomes for degradation discovery of Fzo. Mammalian spermatogenesis is one of the most and recycling of the engulfed organelles. Much of the molecular complex and lengthy differentiation processes in biology, transforming workings of mitophagy have been parsed in cultured cells, and spermatogonial stem cells into highly specialized sperm cells over a recently developed mouse models are enabling the analysis of 5-week period. -

A Network Propagation Approach to Prioritize Long Tail Genes in Cancer
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.05.429983; this version posted February 8, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Network Propagation Approach to Prioritize Long Tail Genes in Cancer Hussein Mohsen1,*, Vignesh Gunasekharan2, Tao Qing2, Sahand Negahban3, Zoltan Szallasi4, Lajos Pusztai2,*, Mark B. Gerstein1,5,6,3,* 1 Computational Biology & Bioinformatics Program, Yale University, New Haven, CT 06511, USA 2 Breast Medical Oncology, Yale School of Medicine, New Haven, CT 06511, USA 3 Department of Statistics & Data Science, Yale University, New Haven, CT 06511, USA 4 Children’s Hospital Informatics Program, Harvard-MIT Division of Health Sciences and Technology, Harvard Medical School, Boston, MA 02115, USA 5 Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511, USA 6 Department of Computer Science, Yale University, New Haven, CT 06511, USA * Corresponding author Abstract Introduction. The diversity of genomic alterations in cancer pose challenges to fully understanding the etiologies of the disease. Recent interest in infrequent mutations, in genes that reside in the “long tail” of the mutational distribution, uncovered new genes with significant implication in cancer development. The study of these genes often requires integrative approaches with multiple types of biological data. Network propagation methods have demonstrated high efficacy in uncovering genomic patterns underlying cancer using biological interaction networks. Yet, the majority of these analyses have focused their assessment on detecting known cancer genes or identifying altered subnetworks. -

Protein Interaction Networks and Their Applications to Protein Characterization and Cancer Genes Prediction
Ramón Aragüés Peleato Protein interaction networks and their applications to protein characterization and cancer genes prediction PhD Thesis Barcelona, May 2007 1 The image of the cover shows the happiness protein interaction network (i.e. the protein interaction network for proteins involved in the serotonin pathway) ii Protein Interaction Networks and their Applications to Protein Characterization and Cancer Genes Prediction Ramón Aragüés Peleato Memòria presentada per optar al grau de Doctor en Biologia per la Universitat Pompeu Fabra. Aquesta Tesi Doctoral ha estat realitzada sota la direcció del Dr. Baldo Oliva al Departament de Ciències Experimentals i de la Salut de la Universitat Pompeu Fabra Baldo Oliva Miguel Ramón Aragüés Peleato Barcelona, Maig 2007 The research in this thesis has been carried out at the Structural Bioinformatics Lab (SBI) within the Grup de Recerca en Informàtica Biomèdica at the Parc de Recerca Biomèdica de Barcelona (PRBB). The research carried out in this thesis has been supported by a “Formación de Personal Investigador (FPI)” grant from the Ministerio de Educación y Ciencia awarded to Dr. Baldo Oliva. A mis padres, que me hicieron querer conseguirlo A Natalia, que me ayudó a conseguir hacerlo ii TABLE OF CONTENTS TABLE OF CONTENTS................................................................................................................................... I ACKNOWLEDGEMENTS........................................................................................................................... -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules Natasha L
www.nature.com/scientificreports OPEN A Multi-Omics Interpretable Machine Learning Model Reveals Modes of Action of Small Molecules Natasha L. Patel-Murray1, Miriam Adam2, Nhan Huynh2, Brook T. Wassie2, Pamela Milani2 & Ernest Fraenkel 2,3* High-throughput screening and gene signature analyses frequently identify lead therapeutic compounds with unknown modes of action (MoAs), and the resulting uncertainties can lead to the failure of clinical trials. We developed an approach for uncovering MoAs through an interpretable machine learning model of transcriptomics, epigenomics, metabolomics, and proteomics. Examining compounds with benefcial efects in models of Huntington’s Disease, we found common MoAs for compounds with unrelated structures, connectivity scores, and binding targets. The approach also predicted highly divergent MoAs for two FDA-approved antihistamines. We experimentally validated these efects, demonstrating that one antihistamine activates autophagy, while the other targets bioenergetics. The use of multiple omics was essential, as some MoAs were virtually undetectable in specifc assays. Our approach does not require reference compounds or large databases of experimental data in related systems and thus can be applied to the study of agents with uncharacterized MoAs and to rare or understudied diseases. Unknown modes of action of drug candidates can lead to unpredicted consequences on efectiveness and safety. Computational methods, such as the analysis of gene signatures, and high-throughput experimental methods have accelerated the discovery of lead compounds that afect a specifc target or phenotype1–3. However, these advances have not dramatically changed the rate of drug approvals. Between 2000 and 2015, 86% of drug can- didates failed to earn FDA approval, with toxicity or a lack of efcacy being common reasons for their clinical trial termination4,5. -
![Downloaded from [266]](https://docslib.b-cdn.net/cover/7352/downloaded-from-266-347352.webp)
Downloaded from [266]
Patterns of DNA methylation on the human X chromosome and use in analyzing X-chromosome inactivation by Allison Marie Cotton B.Sc., The University of Guelph, 2005 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in The Faculty of Graduate Studies (Medical Genetics) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2012 © Allison Marie Cotton, 2012 Abstract The process of X-chromosome inactivation achieves dosage compensation between mammalian males and females. In females one X chromosome is transcriptionally silenced through a variety of epigenetic modifications including DNA methylation. Most X-linked genes are subject to X-chromosome inactivation and only expressed from the active X chromosome. On the inactive X chromosome, the CpG island promoters of genes subject to X-chromosome inactivation are methylated in their promoter regions, while genes which escape from X- chromosome inactivation have unmethylated CpG island promoters on both the active and inactive X chromosomes. The first objective of this thesis was to determine if the DNA methylation of CpG island promoters could be used to accurately predict X chromosome inactivation status. The second objective was to use DNA methylation to predict X-chromosome inactivation status in a variety of tissues. A comparison of blood, muscle, kidney and neural tissues revealed tissue-specific X-chromosome inactivation, in which 12% of genes escaped from X-chromosome inactivation in some, but not all, tissues. X-linked DNA methylation analysis of placental tissues predicted four times higher escape from X-chromosome inactivation than in any other tissue. Despite the hypomethylation of repetitive elements on both the X chromosome and the autosomes, no changes were detected in the frequency or intensity of placental Cot-1 holes. -

Eif2b Is a Decameric Guanine Nucleotide Exchange Factor with a G2e2 Tetrameric Core
ARTICLE Received 19 Feb 2014 | Accepted 15 Apr 2014 | Published 23 May 2014 DOI: 10.1038/ncomms4902 OPEN eIF2B is a decameric guanine nucleotide exchange factor with a g2e2 tetrameric core Yuliya Gordiyenko1,2,*, Carla Schmidt1,*, Martin D. Jennings3, Dijana Matak-Vinkovic4, Graham D. Pavitt3 & Carol V. Robinson1 eIF2B facilitates and controls protein synthesis in eukaryotes by mediating guanine nucleotide exchange on its partner eIF2. We combined mass spectrometry (MS) with chemical cross- linking, surface accessibility measurements and homology modelling to define subunit stoichiometry and interactions within eIF2B and eIF2. Although it is generally accepted that eIF2B is a pentamer of five non-identical subunits (a–e), here we show that eIF2B is a decamer. MS and cross-linking of eIF2B complexes allows us to propose a model for the subunit arrangements within eIF2B where the subunit assembly occurs through catalytic g- and e-subunits, with regulatory subunits arranged in asymmetric trimers associated with the core. Cross-links between eIF2 and eIF2B allow modelling of interactions that contribute to nucleotide exchange and its control by eIF2 phosphorylation. Finally, we identify that GTP binds to eIF2Bg, prompting us to propose a multi-step mechanism for nucleotide exchange. 1 Department of Chemistry, Physical and Theoretical Chemistry Laboratory, University of Oxford, South Parks Road, Oxford OX1 3QZ, UK. 2 MRC Laboratory of Molecular Biology, University of Cambridge, Francis Crick Avenue, Cambridge CB2 0QH, UK. 3 Faculty of Life Sciences, Michael Smith Building, University of Manchester, Oxford Road, Manchester M13 9PT, UK. 4 Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. -

PQBP1, a Factor Linked to Intellectual Disability, Affects Alternative Splicing Associated with Neurite Outgrowth
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press PQBP1, a factor linked to intellectual disability, affects alternative splicing associated with neurite outgrowth Qingqing Wang,1 Michael J. Moore,2 Guillaume Adelmant,3,4,5 Jarrod A. Marto,3,4,5 and Pamela A. Silver1,6,7 1Department of Systems Biology, Harvard Medical School, Boston, Massachusetts 02115, USA; 2Laboratory of Molecular Neuro- Oncology, The Rockefeller University, New York, New York 10065, USA; 3Department of Biological Chemistry and Molecular Pharmacology, Harvard Medical School, Boston, Massachusetts 02115, USA; 4Blais Proteomics Center, 5Department of Cancer Biology, Dana-Farber Cancer Institute, Boston, Massachusetts 02215, USA; 6Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts 02115, USA Polyglutamine-binding protein 1 (PQBP1) is a highly conserved protein associated with neurodegenerative disorders. Here, we identify PQBP1 as an alternative messenger RNA (mRNA) splicing (AS) effector capable of influencing splicing of multiple mRNA targets. PQBP1 is associated with many splicing factors, including the key U2 small nuclear ribonucleoprotein (snRNP) component SF3B1 (subunit 1 of the splicing factor 3B [SF3B] protein complex). Loss of functional PQBP1 reduced SF3B1 substrate mRNA association and led to significant changes in AS patterns. Depletion of PQBP1 in primary mouse neurons reduced dendritic outgrowth and altered AS of mRNAs enriched for functions in neuron projection development. Disease-linked PQBP1 mutants were deficient in splicing factor associations and could not complement neurite outgrowth defects. Our results indicate that PQBP1 can affect the AS of multiple mRNAs and indicate specific affected targets whose splice site determination may contribute to the disease phenotype in PQBP1-linked neurological disorders. -

WO 2015/065964 Al 7 May 2015 (07.05.2015) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/065964 Al 7 May 2015 (07.05.2015) W P O P C T (51) International Patent Classification: (74) Agents: KOWALSKI, Thomas, J. et al; Vedder Price C12N 15/90 (2006.01) C12N 15/113 (2010.01) P.C., 1633 Broadway, New York, NY 1001 9 (US). C12N 15/10 (2006.01) C12N 15/63 (2006.01) (81) Designated States (unless otherwise indicated, for every (21) International Application Number: kind of national protection available): AE, AG, AL, AM, PCT/US2014/062558 AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (22) International Filing Date: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, 28 October 2014 (28.10.2014) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (25) Filing Language: English KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (26) Publication Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (30) Priority Data: SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 61/96 1,980 28 October 20 13 (28. 10.20 13) US TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. 61/963,643 9 December 2013 (09. -

Cofactor Dependent Conformational Switching of Gtpases
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector 1704 Biophysical Journal Volume 95 August 2008 1704–1715 Cofactor Dependent Conformational Switching of GTPases Vasili Hauryliuk,*y Sebastian Hansson,z and Ma˚ns Ehrenberg* *Department of Cell and Molecular Biology, Molecular Biology Program, Uppsala University, Uppsala, Sweden; yEngelhardt Institute of Molecular Biology, Russian Academy of Sciences, Moscow, Russia; and zLaboratoire d’Enzymologie et Biochimie Structurales, CNRS, Avenue de la Terrasse, 91198 Gif-sur-Yvette, France ABSTRACT This theoretical work covers structural and biochemical aspects of nucleotide binding and GDP/GTP exchange of GTP hydrolases belonging to the family of small GTPases. Current models of GDP/GTP exchange regulation are often based on two specific assumptions. The first is that the conformation of a GTPase is switched by the exchange of the bound nucleotide from GDP to GTP or vice versa. The second is that GDP/GTP exchange is regulated by a guanine nucleotide exchange factor, which stabilizes a GTPase conformation with low nucleotide affinity. Since, however, recent biochemical and structural data seem to contradict this view, we present a generalized scheme for GTPase action. This novel ansatz accounts for those important cases when conformational switching in addition to guanine nucleotide exchange requires the presence of cofactors, and gives a more nuanced picture of how the nucleotide exchange is regulated. The scheme is also used to discuss some problems of interpretation that may arise when guanine nucleotide exchange mechanisms are inferred from experiments with analogs of GTP, like GDPNP, GDPCP, and GDP g S. -
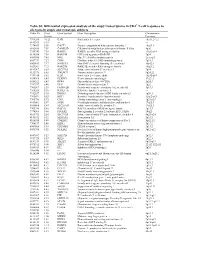
Table S1| Differential Expression Analysis of the Atopy Transcriptome
Table S1| Differential expression analysis of the atopy transcriptome in CD4+ T-cell responses to allergens in atopic and nonatopic subjects Probe ID S.test Gene Symbol Gene Description Chromosome Statistic Location 7994280 10.32 IL4R Interleukin 4 receptor 16p11.2-12.1 8143383 8.95 --- --- --- 7974689 8.50 DACT1 Dapper, antagonist of beta-catenin, homolog 1 14q23.1 8102415 7.59 CAMK2D Calcium/calmodulin-dependent protein kinase II delta 4q26 7950743 7.58 RAB30 RAB30, member RAS oncogene family 11q12-q14 8136580 7.54 RAB19B GTP-binding protein RAB19B 7q34 8043504 7.45 MAL Mal, T-cell differentiation protein 2cen-q13 8087739 7.27 CISH Cytokine inducible SH2-containing protein 3p21.3 8000413 7.17 NSMCE1 Non-SMC element 1 homolog (S. cerevisiae) 16p12.1 8021301 7.15 RAB27B RAB27B, member RAS oncogene family 18q21.2 8143367 6.83 SLC37A3 Solute carrier family 37 member 3 7q34 8152976 6.65 TMEM71 Transmembrane protein 71 8q24.22 7931914 6.56 IL2R Interleukin 2 receptor, alpha 10p15-p14 8014768 6.43 PLXDC1 Plexin domain containing 1 17q21.1 8056222 6.43 DPP4 Dipeptidyl-peptidase 4 (CD26) 2q24.3 7917697 6.40 GFI1 Growth factor independent 1 1p22 7903507 6.39 FAM102B Family with sequence similarity 102, member B 1p13.3 7968236 5.96 RASL11A RAS-like, family 11, member A --- 7912537 5.95 DHRS3 Dehydrogenase/reductase (SDR family) member 3 1p36.1 7963491 5.83 KRT1 Keratin 1 (epidermolytic hyperkeratosis) 12q12-q13 7903786 5.72 CSF1 Colony stimulating factor 1 (macrophage) 1p21-p13 8019061 5.67 SGSH N-sulfoglucosamine sulfohydrolase (sulfamidase) 17q25.3