SLFN11 Promotes CDT1 Degradation by CUL4 in Response to Replicative DNA Damage, While Its Absence Leads to Synthetic Lethality with ATR/CHK1 Inhibitors
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplementary Materials
DEPs in osteosarcoma cells comparing to osteoblastic cells Biological Process Protein Percentage of Hits metabolic process (GO:0008152) 29.3 29.3% cellular process (GO:0009987) 20.2 20.2% localization (GO:0051179) 9.4 9.4% biological regulation (GO:0065007) 8 8.0% developmental process (GO:0032502) 7.8 7.8% response to stimulus (GO:0050896) 5.6 5.6% cellular component organization (GO:0071840) 5.6 5.6% multicellular organismal process (GO:0032501) 4.4 4.4% immune system process (GO:0002376) 4.2 4.2% biological adhesion (GO:0022610) 2.7 2.7% apoptotic process (GO:0006915) 1.6 1.6% reproduction (GO:0000003) 0.8 0.8% locomotion (GO:0040011) 0.4 0.4% cell killing (GO:0001906) 0.1 0.1% 100.1% Genes 2179Hits 3870 biological adhesion apoptotic process … reproduction (GO:0000003) , 0.8% (GO:0022610) , 2.7% locomotion (GO:0040011) ,… immune system process cell killing (GO:0001906) , 0.1% (GO:0002376) , 4.2% multicellular organismal process (GO:0032501) , metabolic process 4.4% (GO:0008152) , 29.3% cellular component organization (GO:0071840) , 5.6% response to stimulus (GO:0050896), 5.6% developmental process (GO:0032502) , 7.8% biological regulation (GO:0065007) , 8.0% cellular process (GO:0009987) , 20.2% localization (GO:0051179) , 9. -

Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model
Downloaded from http://www.jimmunol.org/ by guest on September 25, 2021 T + is online at: average * The Journal of Immunology , 34 of which you can access for free at: 2016; 197:1477-1488; Prepublished online 1 July from submission to initial decision 4 weeks from acceptance to publication 2016; doi: 10.4049/jimmunol.1600589 http://www.jimmunol.org/content/197/4/1477 Molecular Profile of Tumor-Specific CD8 Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. Waugh, Sonia M. Leach, Brandon L. Moore, Tullia C. Bruno, Jonathan D. Buhrman and Jill E. Slansky J Immunol cites 95 articles Submit online. Every submission reviewed by practicing scientists ? is published twice each month by Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts http://jimmunol.org/subscription Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html http://www.jimmunol.org/content/suppl/2016/07/01/jimmunol.160058 9.DCSupplemental This article http://www.jimmunol.org/content/197/4/1477.full#ref-list-1 Information about subscribing to The JI No Triage! Fast Publication! Rapid Reviews! 30 days* Why • • • Material References Permissions Email Alerts Subscription Supplementary The Journal of Immunology The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2016 by The American Association of Immunologists, Inc. All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. This information is current as of September 25, 2021. The Journal of Immunology Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model Katherine A. -

A Network Propagation Approach to Prioritize Long Tail Genes in Cancer
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.05.429983; this version posted February 8, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. A Network Propagation Approach to Prioritize Long Tail Genes in Cancer Hussein Mohsen1,*, Vignesh Gunasekharan2, Tao Qing2, Sahand Negahban3, Zoltan Szallasi4, Lajos Pusztai2,*, Mark B. Gerstein1,5,6,3,* 1 Computational Biology & Bioinformatics Program, Yale University, New Haven, CT 06511, USA 2 Breast Medical Oncology, Yale School of Medicine, New Haven, CT 06511, USA 3 Department of Statistics & Data Science, Yale University, New Haven, CT 06511, USA 4 Children’s Hospital Informatics Program, Harvard-MIT Division of Health Sciences and Technology, Harvard Medical School, Boston, MA 02115, USA 5 Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT 06511, USA 6 Department of Computer Science, Yale University, New Haven, CT 06511, USA * Corresponding author Abstract Introduction. The diversity of genomic alterations in cancer pose challenges to fully understanding the etiologies of the disease. Recent interest in infrequent mutations, in genes that reside in the “long tail” of the mutational distribution, uncovered new genes with significant implication in cancer development. The study of these genes often requires integrative approaches with multiple types of biological data. Network propagation methods have demonstrated high efficacy in uncovering genomic patterns underlying cancer using biological interaction networks. Yet, the majority of these analyses have focused their assessment on detecting known cancer genes or identifying altered subnetworks. -

The Oviductal Extracellular Vesicles' RNA Cargo Regulates the Bovine
International Journal of Molecular Sciences Article The Oviductal Extracellular Vesicles’ RNA Cargo Regulates the Bovine Embryonic Transcriptome Stefan Bauersachs 1 , Pascal Mermillod 2 and Carmen Almiñana 1,2,* 1 Genetics and Functional Genomics, VetSuisse Faculty Zurich, University of Zurich, 8315 Lindau (ZH), Switzerland; [email protected] 2 UMR85 PRC, INRA, CNRS 7247, Université de Tours, IFCE, 37380 Nouzilly, France; [email protected] * Correspondence: [email protected] Received: 21 January 2020; Accepted: 12 February 2020; Published: 14 February 2020 Abstract: Oviductal extracellular vesicles (oEVs) are emerging as key players in the gamete/embryo–oviduct interactions that contribute to successful pregnancy. Various positive effects of oEVs on gametes and early embryos have been found in vitro. To determine whether these effects are associated with changes of embryonic gene expression, the transcriptomes of embryos supplemented with bovine fresh (FeEVs) or frozen (FoEVs) oEVs during in vitro culture compared to controls without oEVs were analyzed by low-input RNA sequencing. Analysis of RNA-seq data revealed 221 differentially expressed genes (DEGs) between FoEV treatment and control, 67 DEGs for FeEV and FoEV treatments, and minor differences between FeEV treatment and control (28 DEGs). An integrative analysis of mRNAs and miRNAs contained in oEVs obtained in a previous study with embryonic mRNA alterations pointed to direct effects of oEV cargo on embryos (1) by increasing the concentration of delivered transcripts; (2) by translating delivered mRNAs to proteins that regulate embryonic gene expression; and (3) by oEV-derived miRNAs which downregulate embryonic mRNAs or modify gene expression in other ways. Our study provided the first high-throughput analysis of the embryonic transcriptome regulated by oEVs, increasing our knowledge on the impact of oEVs on the embryo and revealing the oEV RNA components that potentially regulate embryonic development. -

Protein Interaction Networks and Their Applications to Protein Characterization and Cancer Genes Prediction
Ramón Aragüés Peleato Protein interaction networks and their applications to protein characterization and cancer genes prediction PhD Thesis Barcelona, May 2007 1 The image of the cover shows the happiness protein interaction network (i.e. the protein interaction network for proteins involved in the serotonin pathway) ii Protein Interaction Networks and their Applications to Protein Characterization and Cancer Genes Prediction Ramón Aragüés Peleato Memòria presentada per optar al grau de Doctor en Biologia per la Universitat Pompeu Fabra. Aquesta Tesi Doctoral ha estat realitzada sota la direcció del Dr. Baldo Oliva al Departament de Ciències Experimentals i de la Salut de la Universitat Pompeu Fabra Baldo Oliva Miguel Ramón Aragüés Peleato Barcelona, Maig 2007 The research in this thesis has been carried out at the Structural Bioinformatics Lab (SBI) within the Grup de Recerca en Informàtica Biomèdica at the Parc de Recerca Biomèdica de Barcelona (PRBB). The research carried out in this thesis has been supported by a “Formación de Personal Investigador (FPI)” grant from the Ministerio de Educación y Ciencia awarded to Dr. Baldo Oliva. A mis padres, que me hicieron querer conseguirlo A Natalia, que me ayudó a conseguir hacerlo ii TABLE OF CONTENTS TABLE OF CONTENTS................................................................................................................................... I ACKNOWLEDGEMENTS........................................................................................................................... -

Using Cellminer 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60 William C. Reinhold1, Margot Sunshine1,2, Sudhir
Author Manuscript Published OnlineFirst on June 5, 2015; DOI: 10.1158/1078-0432.CCR-15-0335 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60 William C. Reinhold1, Margot Sunshine1,2, Sudhir Varma1,2,3, James H. Doroshow1,4, and Yves Pommier1 1Developmental Therapeutics Branch and Laboratory of Molecular Pharmacology, Center for Cancer Research, NCI, NIH, Bethesda, Maryland; 2Systems Research and Applications Corp., Fairfax, Virginia; 3HiThru Analytics LLC, Laurel, Maryland; 4Developmental Therapeutics Program, DCTD, NCI, NIH, Bethesda, Maryland. Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/). W.C. Reinhold and Y. Pommier share senior authorship. Corresponding Authors: William C. Reinhold, NIH, 9000 Rockville Pike, Building 37, Room 5041, Bethesda, MD 20892; E-mail: [email protected], and Yves Pommier, NIH, 9000 Rockville Pike, Building 37, Room 5068, Bethesda, MD 20892; E-mail [email protected] Running Title: CellMiner for Systems Pharmacology Disclosure of Potential Conflicts of Interest No potential conflicts of interest were disclosed. Downloaded from clincancerres.aacrjournals.org on September 27, 2021. © 2015 American Association for Cancer Research. Author Manuscript Published OnlineFirst on June 5, 2015; DOI: 10.1158/1078-0432.CCR-15-0335 Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Abstract The NCI-60 cancer cell line panel provides a premier model for data integration and systems pharmacology being the largest publicly available database of anticancer drug activity, , genomic, molecular, and phenotypic data. -

WO 2015/065964 Al 7 May 2015 (07.05.2015) W P O P C T
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (10) International Publication Number (43) International Publication Date WO 2015/065964 Al 7 May 2015 (07.05.2015) W P O P C T (51) International Patent Classification: (74) Agents: KOWALSKI, Thomas, J. et al; Vedder Price C12N 15/90 (2006.01) C12N 15/113 (2010.01) P.C., 1633 Broadway, New York, NY 1001 9 (US). C12N 15/10 (2006.01) C12N 15/63 (2006.01) (81) Designated States (unless otherwise indicated, for every (21) International Application Number: kind of national protection available): AE, AG, AL, AM, PCT/US2014/062558 AO, AT, AU, AZ, BA, BB, BG, BH, BN, BR, BW, BY, BZ, CA, CH, CL, CN, CO, CR, CU, CZ, DE, DK, DM, (22) International Filing Date: DO, DZ, EC, EE, EG, ES, FI, GB, GD, GE, GH, GM, GT, 28 October 2014 (28.10.2014) HN, HR, HU, ID, IL, IN, IR, IS, JP, KE, KG, KN, KP, KR, (25) Filing Language: English KZ, LA, LC, LK, LR, LS, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, (26) Publication Language: English PA, PE, PG, PH, PL, PT, QA, RO, RS, RU, RW, SA, SC, (30) Priority Data: SD, SE, SG, SK, SL, SM, ST, SV, SY, TH, TJ, TM, TN, 61/96 1,980 28 October 20 13 (28. 10.20 13) US TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW. 61/963,643 9 December 2013 (09. -

Supplementary Materials
Supplementary Materials COMPARATIVE ANALYSIS OF THE TRANSCRIPTOME, PROTEOME AND miRNA PROFILE OF KUPFFER CELLS AND MONOCYTES Andrey Elchaninov1,3*, Anastasiya Lokhonina1,3, Maria Nikitina2, Polina Vishnyakova1,3, Andrey Makarov1, Irina Arutyunyan1, Anastasiya Poltavets1, Evgeniya Kananykhina2, Sergey Kovalchuk4, Evgeny Karpulevich5,6, Galina Bolshakova2, Gennady Sukhikh1, Timur Fatkhudinov2,3 1 Laboratory of Regenerative Medicine, National Medical Research Center for Obstetrics, Gynecology and Perinatology Named after Academician V.I. Kulakov of Ministry of Healthcare of Russian Federation, Moscow, Russia 2 Laboratory of Growth and Development, Scientific Research Institute of Human Morphology, Moscow, Russia 3 Histology Department, Medical Institute, Peoples' Friendship University of Russia, Moscow, Russia 4 Laboratory of Bioinformatic methods for Combinatorial Chemistry and Biology, Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry of the Russian Academy of Sciences, Moscow, Russia 5 Information Systems Department, Ivannikov Institute for System Programming of the Russian Academy of Sciences, Moscow, Russia 6 Genome Engineering Laboratory, Moscow Institute of Physics and Technology, Dolgoprudny, Moscow Region, Russia Figure S1. Flow cytometry analysis of unsorted blood sample. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S2. Flow cytometry analysis of unsorted liver stromal cells. Representative forward, side scattering and histogram are shown. The proportions of negative cells were determined in relation to the isotype controls. The percentages of positive cells are indicated. The blue curve corresponds to the isotype control. Figure S3. MiRNAs expression analysis in monocytes and Kupffer cells. Full-length of heatmaps are presented. -

Molecular Pharmacology of Cancer Therapy in Human Colorectal Cancer by Gene Expression Profiling1,2
[CANCER RESEARCH 63, 6855–6863, October 15, 2003] Molecular Pharmacology of Cancer Therapy in Human Colorectal Cancer by Gene Expression Profiling1,2 Paul A. Clarke,3 Mark L. George, Sandra Easdale, David Cunningham, R. Ian Swift, Mark E. Hill, Diana M. Tait, and Paul Workman Cancer Research UK Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, Surrey SM2 5NG [P. A. C., M. L. G., S. E., P. W.]; Department of Gastrointestinal Oncology, Royal Marsden Hospital, Sutton, Surrey [D. C., M. E. H., D. M. T.]; and Department of Surgery, Mayday Hospital, Croydon, Surrey [M. L. G., R. I. S.], United Kingdom ABSTRACT ment with a single dose of MMC4 and during a continuous infusion of 5FU. In this study, we report for the first time gene expression Global gene expression profiling has potential for elucidating the com- profiling in cancer patients before, and critically, during the period of plex cellular effects and mechanisms of action of novel targeted anticancer exposure to chemotherapy. We have demonstrated that the approach agents or existing chemotherapeutics for which the precise molecular is feasible, and we have detected a novel molecular response that mechanism of action may be unclear. In this study, decreased expression would not have been predicted from in vitro studies and that would of genes required for RNA and protein synthesis, and for metabolism were have otherwise been missed by conventional approaches. The results detected in rectal cancer biopsies taken from patients during a 5-fluorou- also suggest a possible new therapeutic approach. Overall our obser- racil infusion. Our observations demonstrate that this approach is feasible and can detect responses that may have otherwise been missed by con- vations suggest that gene expression profiling in response to treatment ventional methods. -

Micrornas Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins
cells Review microRNAs Mediated Regulation of the Ribosomal Proteins and Its Consequences on the Global Translation of Proteins Abu Musa Md Talimur Reza 1,2 and Yu-Guo Yuan 1,3,* 1 Jiangsu Co-Innovation Center of Prevention and Control of Important Animal Infectious Diseases and Zoonoses, College of Veterinary Medicine, Yangzhou University, Yangzhou 225009, China; [email protected] 2 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Pawi´nskiego5a, 02-106 Warsaw, Poland 3 Jiangsu Key Laboratory of Zoonosis/Joint International Research Laboratory of Agriculture and Agri-Product Safety, The Ministry of Education of China, Yangzhou University, Yangzhou 225009, China * Correspondence: [email protected]; Tel.: +86-514-8797-9228 Abstract: Ribosomal proteins (RPs) are mostly derived from the energy-consuming enzyme families such as ATP-dependent RNA helicases, AAA-ATPases, GTPases and kinases, and are important structural components of the ribosome, which is a supramolecular ribonucleoprotein complex, composed of Ribosomal RNA (rRNA) and RPs, coordinates the translation and synthesis of proteins with the help of transfer RNA (tRNA) and other factors. Not all RPs are indispensable; in other words, the ribosome could be functional and could continue the translation of proteins instead of lacking in some of the RPs. However, the lack of many RPs could result in severe defects in the biogenesis of ribosomes, which could directly influence the overall translation processes and global expression of the proteins leading to the emergence of different diseases including cancer. While microRNAs (miRNAs) are small non-coding RNAs and one of the potent regulators of the post-transcriptional 0 gene expression, miRNAs regulate gene expression by targeting the 3 untranslated region and/or coding region of the messenger RNAs (mRNAs), and by interacting with the 50 untranslated region, Citation: Reza, A.M.M.T.; Yuan, Y.-G. -

A Common Analgesic Enhances the Anti-Tumour Activity of 5-Aza-2’- Deoxycytidine Through Induction of Oxidative Stress
bioRxiv preprint doi: https://doi.org/10.1101/2020.03.31.017947; this version posted April 1, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. A common analgesic enhances the anti-tumour activity of 5-aza-2’- deoxycytidine through induction of oxidative stress Hannah J. Gleneadie1,10, Amy H. Baker1, Nikolaos Batis2, Jennifer Bryant2, Yao Jiang3, Samuel J.H. Clokie4, Hisham Mehanna2, Paloma Garcia5, Deena M.A. Gendoo6, Sally Roberts5, Alfredo A. Molinolo7, J. Silvio Gutkind8, Ben A. Scheven1, Paul R. Cooper1, Farhat L. Khanim9 and Malgorzata Wiench1, 5,*. 1School of Dentistry, Institute of Clinical Studies, College of Medical and Dental Sciences, The University of Birmingham, Birmingham, B5 7EG, UK; 2Institute of Head and Neck Studies and Education (InHANSE), The University of Birmingham, Birmingham, B15 2TT, UK; 3School of Biosciences, The University of Birmingham, Birmingham, B15 2TT, UK; 4West Midlands Regional Genetics Laboratory, Birmingham Women’s and Children’s Hospital, Birmingham, B15 2TG, UK; 5Institute of Cancer and Genomic Sciences, College of Medical and Dental Sciences, The University of Birmingham, Birmingham, B15 2TT, UK; 6Centre for Computational Biology, Institute of Cancer and Genomic Sciences, The University of Birmingham, Birmingham, B15 2TT, UK; 7Moores Cancer Center and Department of Pathology, University of California San Diego, La Jolla, CA 92093, USA; 8Department of Pharmacology and Moores Cancer -
Attachment PDF Icon
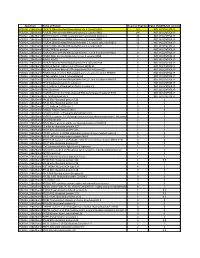
Spectrum Name of Protein Count of Peptides Ratio (POL2RA/IgG control) POLR2A_228kdBand POLR2A DNA-directed RNA polymerase II subunit RPB1 197 NOT IN CONTROL IP POLR2A_228kdBand POLR2B DNA-directed RNA polymerase II subunit RPB2 146 NOT IN CONTROL IP POLR2A_228kdBand RPAP2 Isoform 1 of RNA polymerase II-associated protein 2 24 NOT IN CONTROL IP POLR2A_228kdBand POLR2G DNA-directed RNA polymerase II subunit RPB7 23 NOT IN CONTROL IP POLR2A_228kdBand POLR2H DNA-directed RNA polymerases I, II, and III subunit RPABC3 19 NOT IN CONTROL IP POLR2A_228kdBand POLR2C DNA-directed RNA polymerase II subunit RPB3 17 NOT IN CONTROL IP POLR2A_228kdBand POLR2J RPB11a protein 7 NOT IN CONTROL IP POLR2A_228kdBand POLR2E DNA-directed RNA polymerases I, II, and III subunit RPABC1 8 NOT IN CONTROL IP POLR2A_228kdBand POLR2I DNA-directed RNA polymerase II subunit RPB9 9 NOT IN CONTROL IP POLR2A_228kdBand ALMS1 ALMS1 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2D DNA-directed RNA polymerase II subunit RPB4 6 NOT IN CONTROL IP POLR2A_228kdBand GRINL1A;Gcom1 Isoform 12 of Protein GRINL1A 6 NOT IN CONTROL IP POLR2A_228kdBand RECQL5 Isoform Beta of ATP-dependent DNA helicase Q5 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2L DNA-directed RNA polymerases I, II, and III subunit RPABC5 5 NOT IN CONTROL IP POLR2A_228kdBand KRT6A Keratin, type II cytoskeletal 6A 3 NOT IN CONTROL IP POLR2A_228kdBand POLR2K DNA-directed RNA polymerases I, II, and III subunit RPABC4 2 NOT IN CONTROL IP POLR2A_228kdBand RFC4 Replication factor C subunit 4 1 NOT IN CONTROL IP POLR2A_228kdBand RFC2