Host Species and Body Site Explain the Variation in the Microbiota Associated to Wild 2 Sympatric Mediterranean Teleost Fishes
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The 2014 Golden Gate National Parks Bioblitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event
National Park Service U.S. Department of the Interior Natural Resource Stewardship and Science The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 ON THIS PAGE Photograph of BioBlitz participants conducting data entry into iNaturalist. Photograph courtesy of the National Park Service. ON THE COVER Photograph of BioBlitz participants collecting aquatic species data in the Presidio of San Francisco. Photograph courtesy of National Park Service. The 2014 Golden Gate National Parks BioBlitz - Data Management and the Event Species List Achieving a Quality Dataset from a Large Scale Event Natural Resource Report NPS/GOGA/NRR—2016/1147 Elizabeth Edson1, Michelle O’Herron1, Alison Forrestel2, Daniel George3 1Golden Gate Parks Conservancy Building 201 Fort Mason San Francisco, CA 94129 2National Park Service. Golden Gate National Recreation Area Fort Cronkhite, Bldg. 1061 Sausalito, CA 94965 3National Park Service. San Francisco Bay Area Network Inventory & Monitoring Program Manager Fort Cronkhite, Bldg. 1063 Sausalito, CA 94965 March 2016 U.S. Department of the Interior National Park Service Natural Resource Stewardship and Science Fort Collins, Colorado The National Park Service, Natural Resource Stewardship and Science office in Fort Collins, Colorado, publishes a range of reports that address natural resource topics. These reports are of interest and applicability to a broad audience in the National Park Service and others in natural resource management, including scientists, conservation and environmental constituencies, and the public. The Natural Resource Report Series is used to disseminate comprehensive information and analysis about natural resources and related topics concerning lands managed by the National Park Service. -

Bacterial Epibiotic Communities of Ubiquitous and Abundant Marine Diatoms Are Distinct in Short- and Long-Term Associations
fmicb-09-02879 December 1, 2018 Time: 14:0 # 1 ORIGINAL RESEARCH published: 04 December 2018 doi: 10.3389/fmicb.2018.02879 Bacterial Epibiotic Communities of Ubiquitous and Abundant Marine Diatoms Are Distinct in Short- and Long-Term Associations Klervi Crenn, Delphine Duffieux and Christian Jeanthon* CNRS, Sorbonne Université, Station Biologique de Roscoff, Adaptation et Diversité en Milieu Marin, Roscoff, France Interactions between phytoplankton and bacteria play a central role in mediating biogeochemical cycling and food web structure in the ocean. The cosmopolitan diatoms Thalassiosira and Chaetoceros often dominate phytoplankton communities in marine systems. Past studies of diatom-bacterial associations have employed community- level methods and culture-based or natural diatom populations. Although bacterial assemblages attached to individual diatoms represents tight associations little is known on their makeup or interactions. Here, we examined the epibiotic bacteria of 436 Thalassiosira and 329 Chaetoceros single cells isolated from natural samples and Edited by: collection cultures, regarded here as short- and long-term associations, respectively. Matthias Wietz, Epibiotic microbiota of single diatom hosts was analyzed by cultivation and by cloning- Alfred Wegener Institut, Germany sequencing of 16S rRNA genes obtained from whole-genome amplification products. Reviewed by: The prevalence of epibiotic bacteria was higher in cultures and dependent of the host Lydia Jeanne Baker, Cornell University, United States species. Culture approaches demonstrated that both diatoms carry distinct bacterial Bryndan Paige Durham, communities in short- and long-term associations. Bacterial epibonts, commonly University of Washington, United States associated with phytoplankton, were repeatedly isolated from cells of diatom collection *Correspondence: cultures but were not recovered from environmental cells. -

Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis
International Journal of Molecular Sciences Review Fatty Acid Diets: Regulation of Gut Microbiota Composition and Obesity and Its Related Metabolic Dysbiosis David Johane Machate 1, Priscila Silva Figueiredo 2 , Gabriela Marcelino 2 , Rita de Cássia Avellaneda Guimarães 2,*, Priscila Aiko Hiane 2 , Danielle Bogo 2, Verônica Assalin Zorgetto Pinheiro 2, Lincoln Carlos Silva de Oliveira 3 and Arnildo Pott 1 1 Graduate Program in Biotechnology and Biodiversity in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] (D.J.M.); [email protected] (A.P.) 2 Graduate Program in Health and Development in the Central-West Region of Brazil, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; pri.fi[email protected] (P.S.F.); [email protected] (G.M.); [email protected] (P.A.H.); [email protected] (D.B.); [email protected] (V.A.Z.P.) 3 Chemistry Institute, Federal University of Mato Grosso do Sul, Campo Grande 79079-900, Brazil; [email protected] * Correspondence: [email protected]; Tel.: +55-67-3345-7416 Received: 9 March 2020; Accepted: 27 March 2020; Published: 8 June 2020 Abstract: Long-term high-fat dietary intake plays a crucial role in the composition of gut microbiota in animal models and human subjects, which affect directly short-chain fatty acid (SCFA) production and host health. This review aims to highlight the interplay of fatty acid (FA) intake and gut microbiota composition and its interaction with hosts in health promotion and obesity prevention and its related metabolic dysbiosis. -

British Journal of Nutrition (2014), 111, 2135–2145 Doi:10.1017/S000711451400021X Q the Authors 2014
Downloaded from British Journal of Nutrition (2014), 111, 2135–2145 doi:10.1017/S000711451400021X q The Authors 2014 https://www.cambridge.org/core Iron supplementation promotes gut microbiota metabolic activity but not colitis markers in human gut microbiota-associated rats Alexandra Dostal1, Christophe Lacroix1*, Van T. Pham1, Michael B. Zimmermann2, . IP address: Christophe Del’homme3, Annick Bernalier-Donadille3 and Christophe Chassard1 1Laboratory of Food Biotechnology, Institute of Food, Nutrition and Health, ETH Zurich, Switzerland 170.106.202.8 2Laboratory of Human Nutrition, Institute of Food, Nutrition and Health, ETH Zurich, Switzerland 3UR454 Microbiology Unit, INRA, Clermont-Ferrand Research Centre, St Gene`s-Champanelle, France (Submitted 17 June 2013 – Final revision received 14 November 2013 – Accepted 14 January 2014 – First published online 21 February 2014) , on 01 Oct 2021 at 15:56:07 Abstract The global prevalence of Fe deficiency is high and a common corrective strategy is oral Fe supplementation, which may affect the commensal gut microbiota and gastrointestinal health. The aim of the present study was to investigate the impact of different dietary Fe concentrations on the gut microbiota and gut health of rats inoculated with human faecal microbiota. Rats (8 weeks old, n 40) were divided into five (n 8 each) groups and fed diets differing only in Fe concentration during an Fe-depletion period (12 weeks) and an , subject to the Cambridge Core terms of use, available at Fe-repletion period (4 weeks) as follows: (1) Fe-sufficient diet throughout the study period; (2) Fe-sufficient diet followed by 70 mg Fe/kg diet; (3) Fe-depleted diet throughout the study period; (4) Fe-depleted diet followed by 35 mg Fe/kg diet; (5) Fe-depleted diet followed by 70 mg Fe/kg diet. -
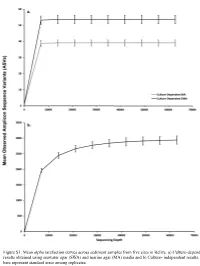
Figure S1. Mean Alpha Rarefaction Curves Across Sediment Samples from Five Sites in Belize
Figure S1. Mean alpha rarefaction curves across sediment samples from five sites in Belize. a) Culture-dependent results obtained using seawater agar (SWA) and marine agar (MA) media and b) Culture- independent results. Error bars represent standard error among replicates. a. 200 150 100 Faith’s PD Faith’s 50 0 Culturedependent MA Culturedependent SWA Cultureindependent Method b. 0.8 0.6 Pielou’s Evenness 0.4 Culturedependent MA Culturedependent SWA Cultureindependent Method Figure S2. Alpha diversity boxplots of marine sediment microbial communities from Carrie Bow Cay, Belize in culture-dependent and culture-independent samples determined using a) Faith’s Phylogenetic Diversity Index and b) Pielou’s Evenness. Culture-dependent methods include the use of marine agar medium (MA) and seawater agar medium (SWA. Data points are overlayed on the boxplot to show variation. a. 100% Proteobacteria Nitrospinae Bacteroidetes Fibrobacteres Planctomycetes Entotheonellaeota 90% Cyanobacteria Marinimicrobia (SAR406 clade) Firmicutes Deinococcus-Thermus Chloroflexi Margulisbacteria [A] Thaumarchaeota Unassigned 80% Acidobacteria Archaea UA Verrucomicrobia Acetothermia Actinobacteria Elusimicrobia 70% [A] Nanoarchaeaeota Tenericutes Kiritimatiellaeota TA06 Latescibacteria WS2 60% Spirochaetes Dependentiae Patescibacteria LCP-89 Gemmatimonadetes FCPU426 Omnitrophicaeota WPS-2 50% Fusobacteria CK-2C2-2 Bacteria UA Armatimonadetes [A] Euryarchaeota [A] Altiarchaeota Relative Percent 40% Calditrichaeota Poribacteria Lentisphaerae Cloacimonetes [A] Crenarchaeota -

Modulation of the Gut Microbiota Alters the Tumour-Suppressive Efficacy of Tim-3 Pathway Blockade in a Bacterial Species- and Host Factor-Dependent Manner
microorganisms Article Modulation of the Gut Microbiota Alters the Tumour-Suppressive Efficacy of Tim-3 Pathway Blockade in a Bacterial Species- and Host Factor-Dependent Manner Bokyoung Lee 1,2, Jieun Lee 1,2, Min-Yeong Woo 1,2, Mi Jin Lee 1, Ho-Joon Shin 1,2, Kyongmin Kim 1,2 and Sun Park 1,2,* 1 Department of Microbiology, Ajou University School of Medicine, Youngtongku Wonchondong San 5, Suwon 442-749, Korea; [email protected] (B.L.); [email protected] (J.L.); [email protected] (M.-Y.W.); [email protected] (M.J.L.); [email protected] (H.-J.S.); [email protected] (K.K.) 2 Department of Biomedical Sciences, The Graduate School, Ajou University, Youngtongku Wonchondong San 5, Suwon 442-749, Korea * Correspondence: [email protected]; Tel.: +82-31-219-5070 Received: 22 August 2020; Accepted: 9 September 2020; Published: 11 September 2020 Abstract: T cell immunoglobulin and mucin domain-containing protein-3 (Tim-3) is an immune checkpoint molecule and a target for anti-cancer therapy. In this study, we examined whether gut microbiota manipulation altered the anti-tumour efficacy of Tim-3 blockade. The gut microbiota of mice was manipulated through the administration of antibiotics and oral gavage of bacteria. Alterations in the gut microbiome were analysed by 16S rRNA gene sequencing. Gut dysbiosis triggered by antibiotics attenuated the anti-tumour efficacy of Tim-3 blockade in both C57BL/6 and BALB/c mice. Anti-tumour efficacy was restored following oral gavage of faecal bacteria even as antibiotic administration continued. In the case of oral gavage of Enterococcus hirae or Lactobacillus johnsonii, transferred bacterial species and host mouse strain were critical determinants of the anti-tumour efficacy of Tim-3 blockade. -

Flavobacterial Response to Organic Pollution
Vol. 51: 31–43, 2008 AQUATIC MICROBIAL ECOLOGY Published April 24 doi: 10.3354/ame01174 Aquat Microb Ecol Flavobacterial response to organic pollution Andrew Bissett1, 2, 3,*, John P. Bowman2, Chris M. Burke1 1School of Aquaculture, Tasmanian Aquaculture and Fisheries Institute, University of Tasmania and Aquafin CRC, Launceston, Tasmania 7250, Australia 2School of Agricultural Science, University of Tasmania, Hobart, Tasmania 7001, Australia 3Present address: Max Planck Institute for Marine Microbiology, Celsiusstrasse 1, 28359 Bremen, Germany ABSTRACT: Bacteria of the Cytophaga-Flavobacterium-Bacteroides (CFB) group (phylum Bac- teroidetes), in particular members of the class Flavobacteria, are among the most prominent hetero- trophic organisms in marine pelagic systems. They have also previously been found to be important in the initial biopolymer degradation of sedimentary organic matter. The Flavobacteria community was analysed in inshore, marine sediments subject to regular inputs of highly labile organic carbon in order to understand the importance of this group in carbon degradation. We used denaturing gra- dient gel electrophoresis and real-time PCR in a statistically robust manner, over 2 consecutive years, to demonstrate that the number of Flavobacteria in the sediment increased and community composi- tion shifted with organic loading. Further community shifts occurred after cessation of organic load- ing, and population numbers also decreased. Flavobacteria appear to be important in the initial responses of the sediment microbial community to organic loading, regardless of sediment type, but flavobacterial composition was not predictable. The highly dynamic nature and large diversity (func- tional redundancy) of the Flavobacteria in these sediments may contribute to this unpredictable response. KEY WORDS: Flavobacteria . -

Individuality in Gut Microbiota Composition Is a Complex Polygenic Trait Shaped by Multiple Environmental and Host Genetic Factors
Individuality in gut microbiota composition is a complex polygenic trait shaped by multiple environmental and host genetic factors Andrew K. Bensona,1, Scott A. Kellyb, Ryan Leggea, Fangrui Maa, Soo Jen Lowa, Jaehyoung Kima, Min Zhanga, Phaik Lyn Oha, Derrick Nehrenbergb, Kunjie Huab, Stephen D. Kachmanc, Etsuko N. Moriyamad, Jens Waltera, Daniel A. Petersona, and Daniel Pompb,e aDepartment of Food Science and Technology and Core for Applied Genomics and Ecology, University of Nebraska, Lincoln, NE 68583-0919; bDepartment of Genetics, Carolina Center for Genome Science, University of North Carolina, Chapel Hill, NC 27599-7545; cDepartment of Statistics, University of Nebraska, Lincoln, NE 68583-0963; dSchool of Biological Sciences and Center for Plant Science Innovation, University of Nebraska, Lincoln, NE 68588-0118; and eDepartment of Nutrition, Gillings School of Global Public Health, University of North Carolina, Chapel Hill, NC 27599-7461 Edited by Trudy F. C. Mackay, North Carolina State University, Raleigh, NC, and approved September 8, 2010 (received for review June 10, 2010) In vertebrates, including humans, individuals harbor gut microbial to be multifactorial, with both environmental and genetic compo- communities whose species composition and relative proportions nents (11–13), and the contribution of the gut microbiota is cur- of dominant microbial groups are tremendously varied. Although rently viewed as an environmental factor (14). Although a number external and stochastic factors clearly contribute to the individu- of studies have suggested that composition of the gut microbiota ality of the microbiota, the fundamental principles dictating how may be subject to host genetic forces, existing evidence is conflicting environmental factors and host genetic factors combine to shape and confounded by the genetic diversity of vertebrate (especially this complex ecosystem are largely unknown and require system- human) populations and strong environmental effects (15–19). -

Compile.Xlsx
Silva OTU GS1A % PS1B % Taxonomy_Silva_132 otu0001 0 0 2 0.05 Bacteria;Acidobacteria;Acidobacteria_un;Acidobacteria_un;Acidobacteria_un;Acidobacteria_un; otu0002 0 0 1 0.02 Bacteria;Acidobacteria;Acidobacteriia;Solibacterales;Solibacteraceae_(Subgroup_3);PAUC26f; otu0003 49 0.82 5 0.12 Bacteria;Acidobacteria;Aminicenantia;Aminicenantales;Aminicenantales_fa;Aminicenantales_ge; otu0004 1 0.02 7 0.17 Bacteria;Acidobacteria;AT-s3-28;AT-s3-28_or;AT-s3-28_fa;AT-s3-28_ge; otu0005 1 0.02 0 0 Bacteria;Acidobacteria;Blastocatellia_(Subgroup_4);Blastocatellales;Blastocatellaceae;Blastocatella; otu0006 0 0 2 0.05 Bacteria;Acidobacteria;Holophagae;Subgroup_7;Subgroup_7_fa;Subgroup_7_ge; otu0007 1 0.02 0 0 Bacteria;Acidobacteria;ODP1230B23.02;ODP1230B23.02_or;ODP1230B23.02_fa;ODP1230B23.02_ge; otu0008 1 0.02 15 0.36 Bacteria;Acidobacteria;Subgroup_17;Subgroup_17_or;Subgroup_17_fa;Subgroup_17_ge; otu0009 9 0.15 41 0.99 Bacteria;Acidobacteria;Subgroup_21;Subgroup_21_or;Subgroup_21_fa;Subgroup_21_ge; otu0010 5 0.08 50 1.21 Bacteria;Acidobacteria;Subgroup_22;Subgroup_22_or;Subgroup_22_fa;Subgroup_22_ge; otu0011 2 0.03 11 0.27 Bacteria;Acidobacteria;Subgroup_26;Subgroup_26_or;Subgroup_26_fa;Subgroup_26_ge; otu0012 0 0 1 0.02 Bacteria;Acidobacteria;Subgroup_5;Subgroup_5_or;Subgroup_5_fa;Subgroup_5_ge; otu0013 1 0.02 13 0.32 Bacteria;Acidobacteria;Subgroup_6;Subgroup_6_or;Subgroup_6_fa;Subgroup_6_ge; otu0014 0 0 1 0.02 Bacteria;Acidobacteria;Subgroup_6;Subgroup_6_un;Subgroup_6_un;Subgroup_6_un; otu0015 8 0.13 30 0.73 Bacteria;Acidobacteria;Subgroup_9;Subgroup_9_or;Subgroup_9_fa;Subgroup_9_ge; -

Gut Microbiota Predicts Healthy Late-Life Aging in Male Mice
bioRxiv preprint doi: https://doi.org/10.1101/2021.06.22.449472; this version posted June 22, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 Gut Microbiota predicts Healthy Late-life Aging in Male Mice 2 Shanlin Ke1,2, Sarah J. Mitchell3,4, Michael R. MacArthur3,4, Alice E. Kane5, David A. 3 Sinclair5, Emily M. Venable6, Katia S. Chadaideh6, Rachel N. Carmody6, Francine 4 Grodstein1,7, James R. Mitchell4, Yang-Yu Liu1 5 6 1Channing Division of Network Medicine, Brigham and Women’s Hospital and Harvard Medical 7 School, Boston, Massachusetts 02115, USA. 8 2State Key Laboratory of Pig Genetic Improvement and Production Technology, Jiangxi Agricultural 9 University 330045, China. 10 3Department of Molecular Metabolism, Harvard T.H. Chan School of Public Health, Boston, MA, 11 02115, USA. 12 4Department of Health Sciences and Technology, ETH Zurich, Zurich 8005 Switzerland. 13 5Blavatnik Institute, Dept. of Genetics, Paul F. Glenn Center for Biology of Aging Research at 14 Harvard Medical School, Boston, MA 02115 USA. 15 6Department of Human Evolutionary Biology, Harvard University, Cambridge, MA, 02138, USA. 16 7Department of Epidemiology, Harvard T.H. Chan School of Public Health, Boston, MA, 02115, USA. 17 18 #To whom correspondence should be addressed: Y.-Y.L. ([email protected]) and 19 S.J.M. ([email protected]) 20 21 Calorie restriction (CR) extends lifespan and retards age-related chronic diseases in most 22 species. There is growing evidence that the gut microbiota has a pivotal role in host health 23 and age-related pathological conditions. -

Pricia Antarctica Gen. Nov., Sp. Nov., a Member of the Family Flavobacteriaceae, Isolated from Antarctic Intertidal Sediment
International Journal of Systematic and Evolutionary Microbiology (2012), 62, 2218–2223 DOI 10.1099/ijs.0.037515-0 Pricia antarctica gen. nov., sp. nov., a member of the family Flavobacteriaceae, isolated from Antarctic intertidal sediment Yong Yu, Hui-Rong Li, Yin-Xin Zeng, Kun Sun and Bo Chen Correspondence SOA Key Laboratory for Polar Science, Polar Research Institute of China, Shanghai 200136, Yong Yu PR China [email protected] A yellow-coloured, rod-shaped, Gram-reaction- and Gram-staining-negative, non-motile and aerobic bacterium, designated strain ZS1-8T, was isolated from a sample of sandy intertidal sediment collected from the Antarctic coast. Flexirubin-type pigments were absent. In phylogenetic analyses based on 16S rRNA gene sequences, strain ZS1-8T formed a distinct phyletic line and the results indicated that the novel strain should be placed in a new genus within the family Flavobacteriaceae. In pairwise comparisons between strain ZS1-8T and recognized species, the levels of 16S rRNA gene sequence similarity were all ,93.3 %. The strain required + + Ca2 and K ions as well as NaCl for growth. Optimal growth was observed at pH 7.5–8.0, 17–19 6C and with 2–3 % (w/v) NaCl. The major fatty acids were iso-C15 : 1 G, iso-C15 : 0, summed feature 3 (iso-C15 : 0 2-OH and/or C16 : 1v7c), an unknown acid with an equivalent chain-length of 13.565 and iso-C17 : 0 3-OH. The major respiratory quinone was MK-6. The predominant polar lipid was phosphatidylethanolamine. The genomic DNA G+C content was 43.9 mol%. -

Crenn Et Al. 2018.Pdf
Bacterial Epibiotic Communities of Ubiquitous and Abundant Marine Diatoms Are Distinct in Short- and Long-Term Associations Klervi Crenn, Delphine Duffieux, Christian Jeanthon To cite this version: Klervi Crenn, Delphine Duffieux, Christian Jeanthon. Bacterial Epibiotic Communities of Ubiquitous and Abundant Marine Diatoms Are Distinct in Short- and Long-Term Associations. Frontiers in Microbiology, Frontiers Media, 2018, 9, pp.2879. 10.3389/fmicb.2018.02879. hal-02130560 HAL Id: hal-02130560 https://hal.archives-ouvertes.fr/hal-02130560 Submitted on 15 May 2019 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. fmicb-09-02879 December 1, 2018 Time: 14:0 # 1 ORIGINAL RESEARCH published: 04 December 2018 doi: 10.3389/fmicb.2018.02879 Bacterial Epibiotic Communities of Ubiquitous and Abundant Marine Diatoms Are Distinct in Short- and Long-Term Associations Klervi Crenn, Delphine Duffieux and Christian Jeanthon* CNRS, Sorbonne Université, Station Biologique de Roscoff, Adaptation et Diversité en Milieu Marin, Roscoff, France Interactions between phytoplankton and bacteria play a central role in mediating biogeochemical cycling and food web structure in the ocean. The cosmopolitan diatoms Thalassiosira and Chaetoceros often dominate phytoplankton communities in marine systems. Past studies of diatom-bacterial associations have employed community- level methods and culture-based or natural diatom populations.