The Intestinal Bacterial Community and Functional Potential of Litopenaeus Vannamei in the Coastal Areas of China
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Metaproteogenomic Insights Beyond Bacterial Response to Naphthalene
ORIGINAL ARTICLE ISME Journal – Original article Metaproteogenomic insights beyond bacterial response to 5 naphthalene exposure and bio-stimulation María-Eugenia Guazzaroni, Florian-Alexander Herbst, Iván Lores, Javier Tamames, Ana Isabel Peláez, Nieves López-Cortés, María Alcaide, Mercedes V. del Pozo, José María Vieites, Martin von Bergen, José Luis R. Gallego, Rafael Bargiela, Arantxa López-López, Dietmar H. Pieper, Ramón Rosselló-Móra, Jesús Sánchez, Jana Seifert and Manuel Ferrer 10 Supporting Online Material includes Text (Supporting Materials and Methods) Tables S1 to S9 Figures S1 to S7 1 SUPPORTING TEXT Supporting Materials and Methods Soil characterisation Soil pH was measured in a suspension of soil and water (1:2.5) with a glass electrode, and 5 electrical conductivity was measured in the same extract (diluted 1:5). Primary soil characteristics were determined using standard techniques, such as dichromate oxidation (organic matter content), the Kjeldahl method (nitrogen content), the Olsen method (phosphorus content) and a Bernard calcimeter (carbonate content). The Bouyoucos Densimetry method was used to establish textural data. Exchangeable cations (Ca, Mg, K and 10 Na) extracted with 1 M NH 4Cl and exchangeable aluminium extracted with 1 M KCl were determined using atomic absorption/emission spectrophotometry with an AA200 PerkinElmer analyser. The effective cation exchange capacity (ECEC) was calculated as the sum of the values of the last two measurements (sum of the exchangeable cations and the exchangeable Al). Analyses were performed immediately after sampling. 15 Hydrocarbon analysis Extraction (5 g of sample N and Nbs) was performed with dichloromethane:acetone (1:1) using a Soxtherm extraction apparatus (Gerhardt GmbH & Co. -

Bioprospecting from Marine Sediments of New Brunswick, Canada: Exploring the Relationship Between Total Bacterial Diversity and Actinobacteria Diversity
Mar. Drugs 2014, 12, 899-925; doi:10.3390/md12020899 OPEN ACCESS marine drugs ISSN 1660-3397 www.mdpi.com/journal/marinedrugs Article Bioprospecting from Marine Sediments of New Brunswick, Canada: Exploring the Relationship between Total Bacterial Diversity and Actinobacteria Diversity Katherine Duncan 1, Bradley Haltli 2, Krista A. Gill 2 and Russell G. Kerr 1,2,* 1 Department of Biomedical Sciences, University of Prince Edward Island, 550 University Avenue, Charlottetown, PE C1A 4P3, Canada; E-Mail: [email protected] 2 Department of Chemistry, University of Prince Edward Island, 550 University Avenue, Charlottetown, PE C1A 4P3, Canada; E-Mails: [email protected] (B.H.); [email protected] (K.A.G.) * Author to whom correspondence should be addressed; E-Mail: [email protected]; Tel.: +1-902-566-0565; Fax: +1-902-566-7445. Received: 13 November 2013; in revised form: 7 January 2014 / Accepted: 21 January 2014 / Published: 13 February 2014 Abstract: Actinomycetes are an important resource for the discovery of natural products with therapeutic properties. Bioprospecting for actinomycetes typically proceeds without a priori knowledge of the bacterial diversity present in sampled habitats. In this study, we endeavored to determine if overall bacterial diversity in marine sediments, as determined by 16S rDNA amplicon pyrosequencing, could be correlated with culturable actinomycete diversity, and thus serve as a powerful tool in guiding future bioprospecting efforts. Overall bacterial diversity was investigated in eight marine sediments from four sites in New Brunswick, Canada, resulting in over 44,000 high quality sequences (x̄ = 5610 per sample). Analysis revealed all sites exhibited significant diversity (H’ = 5.4 to 6.7). -

Eelgrass Sediment Microbiome As a Nitrous Oxide Sink in Brackish Lake Akkeshi, Japan
Microbes Environ. Vol. 34, No. 1, 13-22, 2019 https://www.jstage.jst.go.jp/browse/jsme2 doi:10.1264/jsme2.ME18103 Eelgrass Sediment Microbiome as a Nitrous Oxide Sink in Brackish Lake Akkeshi, Japan TATSUNORI NAKAGAWA1*, YUKI TSUCHIYA1, SHINGO UEDA1, MANABU FUKUI2, and REIJI TAKAHASHI1 1College of Bioresource Sciences, Nihon University, 1866 Kameino, Fujisawa, 252–0880, Japan; and 2Institute of Low Temperature Science, Hokkaido University, Kita-19, Nishi-8, Kita-ku, Sapporo, 060–0819, Japan (Received July 16, 2018—Accepted October 22, 2018—Published online December 1, 2018) Nitrous oxide (N2O) is a powerful greenhouse gas; however, limited information is currently available on the microbiomes involved in its sink and source in seagrass meadow sediments. Using laboratory incubations, a quantitative PCR (qPCR) analysis of N2O reductase (nosZ) and ammonia monooxygenase subunit A (amoA) genes, and a metagenome analysis based on the nosZ gene, we investigated the abundance of N2O-reducing microorganisms and ammonia-oxidizing prokaryotes as well as the community compositions of N2O-reducing microorganisms in in situ and cultivated sediments in the non-eelgrass and eelgrass zones of Lake Akkeshi, Japan. Laboratory incubations showed that N2O was reduced by eelgrass sediments and emitted by non-eelgrass sediments. qPCR analyses revealed that the abundance of nosZ gene clade II in both sediments before and after the incubation as higher in the eelgrass zone than in the non-eelgrass zone. In contrast, the abundance of ammonia-oxidizing archaeal amoA genes increased after incubations in the non-eelgrass zone only. Metagenome analyses of nosZ genes revealed that the lineages Dechloromonas-Magnetospirillum-Thiocapsa and Bacteroidetes (Flavobacteriia) within nosZ gene clade II were the main populations in the N2O-reducing microbiome in the in situ sediments of eelgrass zones. -

Animal Science and Pastures
DOI: http://doi.org/10.1590/1678-992X-2020-0096 ISSN 1678-992X Research Article Effect of dietary protein and genetic line of Litopenaeus vannamei on its hepatopancreatic microbiota Marcel Martinez-Porchas1 , Francisco Vargas-Albores1 , Ramón Casillas-Hernández2 , Libia Zulema Rodriguez-Anaya3 , Fernando Lares-Villa2 , Dante Magdaleno-Moncayo4 , Jose Reyes Gonzalez-Galaviz3* 1Centro de Investigación en Alimentación y Desarrollo A.C., ABSTRACT: Host genetics and diet can exert an influence on microbiota and, therefore, Carretera Gustavo Enrique Astiazarán Rosas, no. 46, Col. La on feeding efficiency. This study evaluated the effect of genetic line (fast-growth and Victoria – C.P. 83304 – Hermosillo, Sonora – Mexico. high-resistance) in Pacific white shrimp (Litopenaeus vannamei) on the hepatopancreatic 2Instituto Tecnológico de Sonora – Depto. de Ciencias microbiota and its association with the feeding efficiency in shrimp fed with diets containing Agronómicas y Veterinarias – Ciudad Obregón, Sonora – different protein sources. Shrimp (2.08 ± 0.06 g) from each genetic line were fed for 36 Mexico. days with two dietary treatments (animal and vegetable protein). Each of the four groups Animal Science and Pastures 3CONACYT – Instituto Tecnológico de Sonora, Calle 5 de was sampled, and the hepatopancreatic metagenome was amplified using specific primers Febrero 818 Sur, Colonia Centro – C.P. 85000 – Ciudad for the variable V4 region of the 16S rRNA gene. The PCR product was sequenced on the Obregón, Sonora – Mexico. MiSeq platform. Nineteen bacterial phyla were detected, of which Proteobacteria was the 4Centro de Investigación Científica y de Educación Superior most abundant (51.0 – 72.5 %), Bacteroidetes (3.6 – 23.3 %), Firmicutes (4.2 – 13.7 %), de Ensenada – Depto. -

The Subway Microbiome: Seasonal Dynamics and Direct Comparison Of
Gohli et al. Microbiome (2019) 7:160 https://doi.org/10.1186/s40168-019-0772-9 RESEARCH Open Access The subway microbiome: seasonal dynamics and direct comparison of air and surface bacterial communities Jostein Gohli1* , Kari Oline Bøifot1,2, Line Victoria Moen1, Paulina Pastuszek3, Gunnar Skogan1, Klas I. Udekwu4 and Marius Dybwad1,2 Abstract Background: Mass transit environments, such as subways, are uniquely important for transmission of microbes among humans and built environments, and for their ability to spread pathogens and impact large numbers of people. In order to gain a deeper understanding of microbiome dynamics in subways, we must identify variables that affect microbial composition and those microorganisms that are unique to specific habitats. Methods: We performed high-throughput 16S rRNA gene sequencing of air and surface samples from 16 subway stations in Oslo, Norway, across all four seasons. Distinguishing features across seasons and between air and surface were identified using random forest classification analyses, followed by in-depth diversity analyses. Results: There were significant differences between the air and surface bacterial communities, and across seasons. Highly abundant groups were generally ubiquitous; however, a large number of taxa with low prevalence and abundance were exclusively present in only one sample matrix or one season. Among the highly abundant families and genera, we found that some were uniquely so in air samples. In surface samples, all highly abundant groups were also well represented in air samples. This is congruent with a pattern observed for the entire dataset, namely that air samples had significantly higher within-sample diversity. We also observed a seasonal pattern: diversity was higher during spring and summer. -
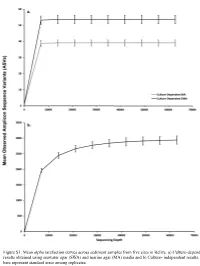
Figure S1. Mean Alpha Rarefaction Curves Across Sediment Samples from Five Sites in Belize
Figure S1. Mean alpha rarefaction curves across sediment samples from five sites in Belize. a) Culture-dependent results obtained using seawater agar (SWA) and marine agar (MA) media and b) Culture- independent results. Error bars represent standard error among replicates. a. 200 150 100 Faith’s PD Faith’s 50 0 Culturedependent MA Culturedependent SWA Cultureindependent Method b. 0.8 0.6 Pielou’s Evenness 0.4 Culturedependent MA Culturedependent SWA Cultureindependent Method Figure S2. Alpha diversity boxplots of marine sediment microbial communities from Carrie Bow Cay, Belize in culture-dependent and culture-independent samples determined using a) Faith’s Phylogenetic Diversity Index and b) Pielou’s Evenness. Culture-dependent methods include the use of marine agar medium (MA) and seawater agar medium (SWA. Data points are overlayed on the boxplot to show variation. a. 100% Proteobacteria Nitrospinae Bacteroidetes Fibrobacteres Planctomycetes Entotheonellaeota 90% Cyanobacteria Marinimicrobia (SAR406 clade) Firmicutes Deinococcus-Thermus Chloroflexi Margulisbacteria [A] Thaumarchaeota Unassigned 80% Acidobacteria Archaea UA Verrucomicrobia Acetothermia Actinobacteria Elusimicrobia 70% [A] Nanoarchaeaeota Tenericutes Kiritimatiellaeota TA06 Latescibacteria WS2 60% Spirochaetes Dependentiae Patescibacteria LCP-89 Gemmatimonadetes FCPU426 Omnitrophicaeota WPS-2 50% Fusobacteria CK-2C2-2 Bacteria UA Armatimonadetes [A] Euryarchaeota [A] Altiarchaeota Relative Percent 40% Calditrichaeota Poribacteria Lentisphaerae Cloacimonetes [A] Crenarchaeota -

Presence of Pacific White Shrimp Litopenaeus Vannamei (Boone, 1931) in the Southern Gulf of Mexico
Aquatic Invasions (2011) Volume 6, Supplement 1: S139–S142 doi: 10.3391/ai.2011.6.S1.031 Open Access © 2011 The Author(s). Journal compilation © 2011 REABIC Aquatic Invasions Records Presence of Pacific white shrimp Litopenaeus vannamei (Boone, 1931) in the Southern Gulf of Mexico Armando T. Wakida-Kusunoki1*, Luis Enrique Amador-del Angel2, Patricia Carrillo Alejandro1 and Cecilia Quiroga Brahms1 1Instituto Nacional de Pesca, Ave. Héroes del 21 de Abril s/n. Col Playa Norte, Ciudad del Carmen Campeche, México 2Universidad Autónoma del Carmen, Centro de Investigación de Ciencias Ambientales (CICA), Ave. Laguna de Términos s/n Col. Renovación 2da Sección, C.P. 24155, Ciudad del Carmen, Campeche, México E-mail: [email protected] (ATWK), [email protected] (LEAA), [email protected] (PCA), [email protected] (CQB) *Corresponding author Received: 12 July 2011 / Accepted: 12 October 2011 / Published online: 27 October 2011 Abstract This is the first report of the presence of Pacific white shrimp Litopenaeus vannamei in the Southern Gulf of Mexico coast. Seven specimens were collected in the Carmen-Pajonal-Machona lagoons near La Azucena and Sanchez Magallanes in Tabasco, Mexico, during a shrimp monitoring program survey conducted in this area. Further sampling and monitoring are required to find evidence that confirms the establishment of a population of Pacific white shrimp L. vannamei in Southern Gulf of Mexico. Key words: Litopenaeus vannamei, Pacific white shrimp, invasive species, Tabasco, Mexico Introduction covering 319.6 ha (Diario Oficial de la Federacion 2011). Almost all of these farms are Litopenaeus vannamei (Boone, 1931) is native to located in the Southern part of the Machona the Eastern Pacific coast from the Gulf of Lagoon. -

Diversity and Taxonomic Novelty of Actinobacteria Isolated from The
Diversity and taxonomic novelty of Actinobacteria isolated from the Atacama Desert and their potential to produce antibiotics Dissertation zur Erlangung des Doktorgrades der Mathematisch-Naturwissenschaftlichen Fakultät der Christian-Albrechts-Universität zu Kiel Vorgelegt von Alvaro S. Villalobos Kiel 2018 Referent: Prof. Dr. Johannes F. Imhoff Korreferent: Prof. Dr. Ute Hentschel Humeida Tag der mündlichen Prüfung: Zum Druck genehmigt: 03.12.2018 gez. Prof. Dr. Frank Kempken, Dekan Table of contents Summary .......................................................................................................................................... 1 Zusammenfassung ............................................................................................................................ 2 Introduction ...................................................................................................................................... 3 Geological and climatic background of Atacama Desert ............................................................. 3 Microbiology of Atacama Desert ................................................................................................. 5 Natural products from Atacama Desert ........................................................................................ 9 References .................................................................................................................................. 12 Aim of the thesis ........................................................................................................................... -

Microbiology in Shale: Alternatives for Enhanced Gas Recovery
Graduate Theses, Dissertations, and Problem Reports 2015 Microbiology in Shale: Alternatives for Enhanced Gas Recovery Yael Tarlovsky Tucker Follow this and additional works at: https://researchrepository.wvu.edu/etd Recommended Citation Tucker, Yael Tarlovsky, "Microbiology in Shale: Alternatives for Enhanced Gas Recovery" (2015). Graduate Theses, Dissertations, and Problem Reports. 6834. https://researchrepository.wvu.edu/etd/6834 This Dissertation is protected by copyright and/or related rights. It has been brought to you by the The Research Repository @ WVU with permission from the rights-holder(s). You are free to use this Dissertation in any way that is permitted by the copyright and related rights legislation that applies to your use. For other uses you must obtain permission from the rights-holder(s) directly, unless additional rights are indicated by a Creative Commons license in the record and/ or on the work itself. This Dissertation has been accepted for inclusion in WVU Graduate Theses, Dissertations, and Problem Reports collection by an authorized administrator of The Research Repository @ WVU. For more information, please contact [email protected]. Microbiology in Shale: Alternatives for Enhanced Gas Recovery Yael Tarlovsky Tucker Dissertation submitted to the Davis College of Agriculture, Natural Resources and Design at West Virginia University in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Genetics and Developmental Biology Jianbo Yao, Ph.D., Chair James Kotcon, Ph.D. -

Flavobacterial Response to Organic Pollution
Vol. 51: 31–43, 2008 AQUATIC MICROBIAL ECOLOGY Published April 24 doi: 10.3354/ame01174 Aquat Microb Ecol Flavobacterial response to organic pollution Andrew Bissett1, 2, 3,*, John P. Bowman2, Chris M. Burke1 1School of Aquaculture, Tasmanian Aquaculture and Fisheries Institute, University of Tasmania and Aquafin CRC, Launceston, Tasmania 7250, Australia 2School of Agricultural Science, University of Tasmania, Hobart, Tasmania 7001, Australia 3Present address: Max Planck Institute for Marine Microbiology, Celsiusstrasse 1, 28359 Bremen, Germany ABSTRACT: Bacteria of the Cytophaga-Flavobacterium-Bacteroides (CFB) group (phylum Bac- teroidetes), in particular members of the class Flavobacteria, are among the most prominent hetero- trophic organisms in marine pelagic systems. They have also previously been found to be important in the initial biopolymer degradation of sedimentary organic matter. The Flavobacteria community was analysed in inshore, marine sediments subject to regular inputs of highly labile organic carbon in order to understand the importance of this group in carbon degradation. We used denaturing gra- dient gel electrophoresis and real-time PCR in a statistically robust manner, over 2 consecutive years, to demonstrate that the number of Flavobacteria in the sediment increased and community composi- tion shifted with organic loading. Further community shifts occurred after cessation of organic load- ing, and population numbers also decreased. Flavobacteria appear to be important in the initial responses of the sediment microbial community to organic loading, regardless of sediment type, but flavobacterial composition was not predictable. The highly dynamic nature and large diversity (func- tional redundancy) of the Flavobacteria in these sediments may contribute to this unpredictable response. KEY WORDS: Flavobacteria . -

Francisca Rodrigues Dos Reis
Universidade do Minho Escola de Ciências Francisca Rodrigues dos Reis Effect of mycorrhization on Quercus suber L. tolerance to drought L. tolerance to drought cus suber Quer corrhization on y fect of m Ef Francisca Rodrigues dos Reis Governo da República Portuguesa UMinho|2018 janeiro de 2018 Universidade do Minho Escola de Ciências Francisca Rodrigues dos Reis Effect of mycorrhization on Quercus suber L. tolerance to drought Tese de Doutoramento Programa Doutoral em Biologia de Plantas Trabalho efetuado sob a orientação da Profª Doutora Teresa Lino-Neto da Profª Doutora Paula Baptista e do Prof. Doutor Rui Tavares janeiro de 2018 Acknowledgements “Em tudo obrigada!” Quando um dia me vi sentada num anfiteatro onde me descreviam a importância da criação de laços e que o sentimento de gratidão deveria estar implícito no nosso dia-a-dia, nunca pensar que seria o mote de início da minha tese de Doutoramento. Quanto mais não seja por ter sido um padre jesuíta a dizer-mo numa reunião de pais. Sim, é verdade! Para quem acompanhou toda a minha jornada sabe que não fiz um doutoramento tradicional, que não sou uma pessoa convencional e que não encaixo nas estatísticas de uma jovem investigadora! Tudo isto me foi proporcionado graças à pessoa mais humana e à orientadora mais presente que poderia ter escolhido. A Profa. Teresa foi muito mais do que uma simples orientadora. Foi quem me incentivou quando a moral andava baixa, foi a disciplinadora quando o entusiasmo era desmedido, foi a amiga nos momentos de desespero. Apanhar amostras sob neve e temperaturas negativas, acordar de madrugada sob olhar atento de veados, e lama, lama e mais lama, são algumas das recordações que vou guardar para a vida! Obrigada por me proporcionar experiências de uma vida! Ao Prof. -

Effect of a Black Soldier Fly Ingredient on the Growth Performance and Disease Resistance of Juvenile Pacific White Shrimp
animals Article Effect of a Black Soldier Fly Ingredient on the Growth Performance and Disease Resistance of Juvenile Pacific White Shrimp (Litopenaeus vannamei) Andrew Richardson 1,*, João Dantas-Lima 2, Maxime Lefranc 1 and Maye Walraven 1 1 Innovafeed SAS, 75010 Paris, France; [email protected] (M.L.); [email protected] (M.W.) 2 IMAQUA, 9090 Lochristi, Belgium; [email protected] * Correspondence: [email protected]; Tel.: +44-7867-384-167 Simple Summary: This study investigates the use of a Black soldier fly (Hermetia illucens) ingredient in juvenile shrimp (Litopenaeus vannamei) diets at various inclusion rates (4.5, 7.5, and 10.5%), monitor- ing both the growth performance and then health performance in the face of three separate challenges (White spot syndrome virus, Vibrio parahaemolyticus, and osmotic stress). This work showed that growth performance (measured through weight gain, feed conversion ratio, and specific growth rate) of L. vannamei was significantly improved in a linear trend with the inclusion of the Black soldier fly ingredient (p < 0.05), whilst health performance was not significantly altered. Overall, the Black soldier fly ingredient proves to be a promising additive for L. vannamei diets, impacting performance and sustainability positively. Citation: Richardson, A.; Dantas-Lima, J.; Lefranc, M.; Abstract: This study was performed as part of developing a functional feed ingredient for juvenile Walraven, M. Effect of a Black Soldier Pacific white shrimp (Litopenaeus vannamei). Here we assess the effects of dietary inclusion of a Fly Ingredient on the Growth Black Soldier Fly Ingredient (BSFI) from defatted black soldier fly (Hermetia illucens) larvae meal Performance and Disease Resistance on growth performance, tolerance to salinity stress, and disease resistance when challenged with of Juvenile Pacific White Shrimp (Litopenaeus vannamei).