B21167564.Pdf
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 ICR-Geneset Gene List
ICR-geneset Gene List. IMAGE ID UniGene Locus Name Cluster 20115 Hs.62185 SLC9A6 solute carrier family 9 (sodium/hydrogen exchanger), isoform 6 21738 21899 Hs.78353 SRPK2 SFRS protein kinase 2 21908 Hs.79133 CDH8 cadherin 8, type 2 22040 Hs.151738 MMP9 matrix metalloproteinase 9 (gelatinase B, 92kD gelatinase, 92kD type IV collagenase) 22411 Hs.183 FY Duffy blood group 22731 Hs.1787 PHRET1 PH domain containing protein in retina 1 22859 Hs.356487 ESTs 22883 Hs.150926 FPGT fucose-1-phosphate guanylyltransferase 22918 Hs.346868 EBNA1BP2 EBNA1 binding protein 2 23012 Hs.158205 BLZF1 basic leucine zipper nuclear factor 1 (JEM-1) 23073 Hs.284244 FGF2 fibroblast growth factor 2 (basic) 23173 Hs.151051 MAPK10 mitogen-activated protein kinase 10 23185 Hs.289114 TNC tenascin C (hexabrachion) 23282 Hs.8024 IK IK cytokine, down-regulator of HLA II 23353 23431 Hs.50421 RB1CC1 RB1-inducible coiled-coil 1 23514 23548 Hs.71848 Human clone 23548 mRNA sequence 23629 Hs.135587 Human clone 23629 mRNA sequence 23658 Hs.265855 SETMAR SET domain and mariner transposase fusion gene 23676 Hs.100841 Homo sapiens clone 23676 mRNA sequence 23772 Hs.78788 LZTR1 leucine-zipper-like transcriptional regulator, 1 23776 Hs.75438 QDPR quinoid dihydropteridine reductase 23804 Hs.343586 ZFP36 zinc finger protein 36, C3H type, homolog (mouse) 23831 Hs.155247 ALDOC aldolase C, fructose-bisphosphate 23878 Hs.99902 OPCML opioid binding protein/cell adhesion molecule-like 23903 Hs.12526 Homo sapiens clone 23903 mRNA sequence 23932 Hs.368063 Human clone 23932 mRNA sequence 24004 -

Download The
PROBING THE INTERACTION OF ASPERGILLUS FUMIGATUS CONIDIA AND HUMAN AIRWAY EPITHELIAL CELLS BY TRANSCRIPTIONAL PROFILING IN BOTH SPECIES by POL GOMEZ B.Sc., The University of British Columbia, 2002 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE in THE FACULTY OF GRADUATE STUDIES (Experimental Medicine) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2010 © Pol Gomez, 2010 ABSTRACT The cells of the airway epithelium play critical roles in host defense to inhaled irritants, and in asthma pathogenesis. These cells are constantly exposed to environmental factors, including the conidia of the ubiquitous mould Aspergillus fumigatus, which are small enough to reach the alveoli. A. fumigatus is associated with a spectrum of diseases ranging from asthma and allergic bronchopulmonary aspergillosis to aspergilloma and invasive aspergillosis. Airway epithelial cells have been shown to internalize A. fumigatus conidia in vitro, but the implications of this process for pathogenesis remain unclear. We have developed a cell culture model for this interaction using the human bronchial epithelium cell line 16HBE and a transgenic A. fumigatus strain expressing green fluorescent protein (GFP). Immunofluorescent staining and nystatin protection assays indicated that cells internalized upwards of 50% of bound conidia. Using fluorescence-activated cell sorting (FACS), cells directly interacting with conidia and cells not associated with any conidia were sorted into separate samples, with an overall accuracy of 75%. Genome-wide transcriptional profiling using microarrays revealed significant responses of 16HBE cells and conidia to each other. Significant changes in gene expression were identified between cells and conidia incubated alone versus together, as well as between GFP positive and negative sorted cells. -

Predicting Clinical Response to Treatment with a Soluble Tnf-Antagonist Or Tnf, Or a Tnf Receptor Agonist
(19) TZZ _ __T (11) EP 2 192 197 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 02.06.2010 Bulletin 2010/22 C12Q 1/68 (2006.01) (21) Application number: 08170119.5 (22) Date of filing: 27.11.2008 (84) Designated Contracting States: (72) Inventor: The designation of the inventor has not AT BE BG CH CY CZ DE DK EE ES FI FR GB GR yet been filed HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT RO SE SI SK TR (74) Representative: Habets, Winand Designated Extension States: Life Science Patents AL BA MK RS PO Box 5096 6130 PB Sittard (NL) (71) Applicant: Vereniging voor Christelijk Hoger Onderwijs, Wetenschappelijk Onderzoek en Patiëntenzorg 1081 HV Amsterdam (NL) (54) Predicting clinical response to treatment with a soluble tnf-antagonist or tnf, or a tnf receptor agonist (57) The invention relates to methods for predicting a clinical response to a therapy with a soluble TNF antagonist, TNF or a TNF receptor agonist and a kit for use in said methods. EP 2 192 197 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 192 197 A1 Description [0001] The invention relates to methods for predicting a clinical response to a treatment with a soluble TNF antagonist, with TNF or a TNF receptor agonist using expression levels of genes of the Type I INF pathway and a kit for use in said 5 methods. In another aspect, the invention relates to a method for evaluating a pharmacological effect of a treatment with a soluble TNF antagonist, TNF or a TNF receptor agonist. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Characterisation of Isomirs in Stem Cells
Characterisation of isomiRs in stem cells Geok Chin Tan Institute of Reproductive and Developmental Biology Department of Surgery and Cancer Faculty of Medicine Imperial College London Thesis submitted to Imperial College London for the degree of Doctor of Philosophy 1 Statement of Originality All experiments included in this thesis were performed by me unless otherwise stated in the text. 2 Copyright Statement ‘The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives licence. Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the licence terms of this work’ 3 Acknowledgements I would like to thank my supervisor Dr Nicholas Dibb for giving me the opportunity to work in his lab and for all of his guidance and support throughout my PhD, without which this project would not have been possible. I am also very grateful to Dr Wei Cui for teaching me the technique of stem cell culture, her comments on my project related to stem cells and as a wonderful co-supervisor. I would like to also thank Professor Malcolm Parker for his supports and advise on academic and non-academic related subjects. Many thanks to Elcie Chan for the generation of all the stem cell libraries which forms the platform for my project. My sincere thanks also to Gunter Meister for supplying the Argonaute antibodies, Leandro Castellano for the help in the design of RNA sponges, Laki Buluwela for the pTRIPz lentiviral vector and last but not least Alywn Dart from Charlotte Bevan group for the prostate cancer cell lines. -
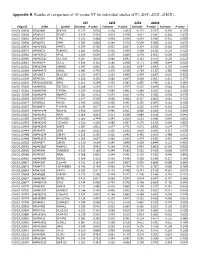
Appendix B. Results of Comparison of AI Versus NT for Individual Studies (D7e, D25e, D25x, D280x)
Appendix B. Results of comparison of AI versus NT for individual studies (d7E, d25E, d25X, d280X). d7E d25E d25X d280X Oligo ID cDNA Symbol Estimate P-value Estimate P-value Estimate P-value Estimate P-value OLIGO_00038 BF042860 RNF166 -0.123 0.634 0.194 0.469 -0.171 0.537 -0.302 0.263 OLIGO_00040 BF045167 POMT1 0.019 0.903 0.010 0.978 0.411 0.181 -0.204 0.476 OLIGO_00043 BF043567 ETFA 0.131 0.484 -0.094 0.706 -0.053 0.745 -0.331 0.056 OLIGO_00068 BF042951 GPX3 0.023 0.840 -0.249 0.370 -0.290 0.088 0.181 0.437 OLIGO_00070 AW464501 HPRT1 -0.079 0.785 0.055 0.837 0.224 0.259 -0.220 0.097 OLIGO_00074 BF040652 PLEKHC1 0.085 0.691 -0.256 0.399 0.309 0.156 0.133 0.423 OLIGO_00081 AW463422 PPIL1 0.109 0.600 0.142 0.686 0.379 0.061 -0.609 0.003 OLIGO_00086 AW462335 SULT1A1 -0.027 0.870 -0.083 0.845 0.123 0.675 0.520 0.121 OLIGO_00087 BF046074 CCL15 -0.306 0.132 -0.585 0.038 -0.771 0.038 0.844 0.020 OLIGO_00161 BM362588 USMG5 0.047 0.836 -0.136 0.628 0.547 0.043 0.279 0.064 OLIGO_00166 AW464473 UFD1L 0.083 0.742 0.372 0.538 0.759 0.132 0.285 0.418 OLIGO_00196 BF039617 BLOC1S2 0.011 0.972 0.153 0.699 0.424 0.187 0.030 0.931 OLIGO_00209 BF045356 GRB2 0.226 0.156 0.685 0.007 -0.008 0.961 0.212 0.177 OLIGO_00220 BP230002B20E6 NFX1 -0.161 0.294 -0.133 0.583 0.055 0.746 -0.211 0.290 OLIGO_00228 AW465641 TBC1D12 -0.248 0.236 -0.013 0.975 -0.223 0.496 0.044 0.821 OLIGO_00261 AW463949 TATDN1 0.020 0.914 -0.089 0.861 -0.248 0.611 -0.412 0.264 OLIGO_00308 AW464976 SNAPC3 0.021 0.911 -0.233 0.417 -0.054 0.782 0.004 0.979 OLIGO_00329 BF045997 PTP4A1 0.247 0.250 -

Ncomms6059.Pdf
ARTICLE Received 4 Feb 2014 | Accepted 21 Aug 2014 | Published 6 Oct 2014 DOI: 10.1038/ncomms6059 OPEN ABRO1 suppresses tumourigenesis and regulates the DNA damage response by stabilizing p53 Jianhong Zhang1,*, Mengmeng Cao2,*, Jiahong Dong3, Changyan Li1, Wangxiang Xu1, Yiqun Zhan1, Xiaohui Wang1, Miao Yu1, Changhui Ge1, Zhiqiang Ge2 & Xiaoming Yang1,2 Abraxas brother 1 (ABRO1) has been reported to be a component of the BRISC complex, a multiprotein complex that specifically cleaves ‘Lys-63’-linked ubiquitin. However, current knowledge of the functions of ABRO1 is limited. Here we report that ABRO1 is frequently downregulated in human liver, kidney, breast and thyroid gland tumour tissues. Depletion of ABRO1 in cancer cells reduces p53 levels and enhances clone formation and cellular transformation. Conversely, overexpression of ABRO1 suppresses cell proliferation and tumour formation in a p53- dependent manner. We further show that ABRO1 stabilizes p53 by facilitating the interaction of p53 with USP7. DNA-damage induced accumulation of endogenous ABRO1 as well as translocation of ABRO1 to the nucleus, and the induction of p53 by DNA damage is almost completely attenuated by ABRO1 depletion. Our study shows that ABRO1 is a novel p53 regulator that plays an important role in tumour suppression and the DNA damage response. 1 State Key Laboratory of Proteomics, Beijing Proteomics Research Center, Beijing Institute of Radiation Medicine, 27-Taiping Road, Beijing 100850, China. 2 School of Chemical Engineering and Technology, TianJin University, Tianjin 300073, China. 3 Department of Liver and Gallbladder Surgery, The General Hospital of Chinese People’s Liberation Army, Beijing 100853, China. * These authors contributed equally to this work. -

Antibodies Products
Chapter 2 : Gentaur Products List • WDSUB1 contains 1 SAM sterile alpha motif domain 1 U Defects in DLG3 are the cause of mental retardation X • Sodium hydrogen exchangers NHEs such as SLC9A8 are box domain and 7 WD repeats The function of WDSUB1 linked type 90 MRX90 integral transmembrane proteins that exchange extracellular remains unknown • SLC25A20 is one of several closely related mitochondrial Na for intracellular H NHEs have multiple functions including • RNF217 is an E3 ubiquitin protein ligase which accepts membrane carrier proteins that shuttle substrates between intracellular pH ho ubiquitin from E2 ubiquitin conjugating enzymes in the form cytosol and the intramitochondrial matrix space It mediates • SLCO1C1 is a member of the organic anion transporter of a thioester and then directly transfers the ubiquitin to the transport of acylcarn family SLCO1C1 is a transmembrane receptor that mediates targeted substrates • The sodium iodide symporter NIS or SLC5A5 is a key the sodium independent uptake of thyroid hormones in brain • TRIM60 contains a RING finger domain a motif present in a plasma membrane protein that mediates active I uptake in tissues This protein has part variety of functionally distinct proteins and known to be thyroid lactating breast and other tissues with an • SLC35A5 belongs to the nucleotide sugar transporter involved in protein protein and protein DNA interactions The electrogenic stoichiometry of 2 Na family SLC35A subfamily It is a multi pass membrane protein encoded by thi • SLC2A9 is a member of the SLC2A -

Supplementary Data
SUPPLEMENTARY METHODS 1) Characterisation of OCCC cell line gene expression profiles using Prediction Analysis for Microarrays (PAM) The ovarian cancer dataset from Hendrix et al (25) was used to predict the phenotypes of the cell lines used in this study. Hendrix et al (25) analysed a series of 103 ovarian samples using the Affymetrix U133A array platform (GEO: GSE6008). This dataset comprises clear cell (n=8), endometrioid (n=37), mucinous (n=13) and serous epithelial (n=41) primary ovarian carcinomas and samples from 4 normal ovaries. To build the predictor, the Prediction Analysis of Microarrays (PAM) package in R environment was employed (http://rss.acs.unt.edu/Rdoc/library/pamr/html/00Index.html). When more than one probe described the expression of a given gene, we used the probe with the highest median absolute deviation across the samples. The dataset from Hendrix et al. (25) and the dataset of OCCC cell lines described in this manuscript were then overlaid on the basis of 11536 common unique HGNC gene symbols. Only the 99 primary ovarian cancers samples and the four normal ovary samples were used to build the predictor. Following leave one out cross-validation, a predictor based upon 126 genes was able to identify correctly the four distinct phenotypes of primary ovarian tumour samples with a misclassification rate of 18.3%. This predictor was subsequently applied to the expression data from the 12 OCCC cell lines to determine the likeliest phenotype of the OCCC cell lines compared to primary ovarian cancers. Posterior probabilities were estimated for each cell line in comparison to the following phenotypes: clear cell, endometrioid, mucinous and serous epithelial. -

Role of Microrna in Early Life Placental Programming of Insulin Resistance and Metabolic Health
Role of MicroRNA in Early Life Placental Programming of Insulin Resistance and Metabolic Health Himawan Harryanto, BSc (Biotechnology), BHSc (Hons) Discipline of Obstetrics and Gynaecology School of Paediatrics and Reproductive Health Faculty of Health Sciences The University of Adelaide South Australia Australia Thesis submitted for the fulfilment for the degree of Doctor of Philosophy (PhD) February 2014 ‘I love fools’ experiment, I am always making them’ – Charles Darwin Table of Contents TABLE OF CONTENTS LIST OF TABLES AND FIGURES ............................................................................. vi ACKNOWLEDGEMENTS ........................................................................................... x STATEMENT OF ORIGINALITY AND AUTHETICITY ...............................................xii TABLE OF ABBREVIATIONS AND BIOCHEMICAL NAMES ................................... xiii CONFERENCE PRESENTATIONS ARISING FROM THIS THESIS ........................xvi ABSTRACT……. ..................................................................................................... xviii Chapter 1 Introduction ................................................................................................. 1 1.1 Overview of the thesis .................................................................................... 3 1.2 IUGR and subsequent metabolic disease ...................................................... 6 1.2.1 Consequences of IUGR ........................................................................... 6 1.2.2 Type -
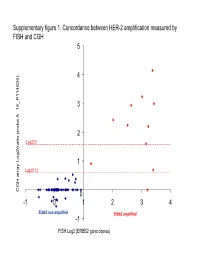
Supplementary Data
Supplementary figure 1. Concordance between HER-2 amplification measured by FISH and CGH 5 4 6) 3 14_P11482 __ 2 Log2(3) atio probe A atio probe rr 1 Log2(1.5) array Log2( CGH 0 -1 0 1 2 3 4 Er bb2 non amplifi e d Erbb2 amplified -1 FISH Log2 (ERBB2 gene copies) Supplementary Figure 2. Gained and lost regions according to triple-negativity Non-triple negative tumors (n=69) Triple negative tumors (n=35) Non-triple negative tumors (n=69) vs triple negative tumors (n=35) Supplementary Figure 3. Gained and lost regions according to chemotherapy response Supplementary Figure 4. Gained and lost regions according to taxanes efficacy Supplementary Table 1. Patient characteristics (n=106). Characteristic N (%) Age (median, range) 51 (28-73) cT cT0/T1 6 (6%) cT2 53 (50%) cT3 18 (17%) cT4 29 (17%) cN cN0 24 (23%) cN1 50 (47%) cN2 23 (22%) cN3 9 (8%) Tumor grade Grade 1 6 (6%) Grade 2 39 (37%) Grade 3 47 (43%) Not assessable 14 (14%) Estrogen receptor expression Negative 45 (42%) Positive 60 (57%) Unknown 1 (1%) Progesteron receptor expression Negative 57 (54%) Positive 47 (44%) Unknown 2 (2%) Her2 status Normal 95 (90%) Overexpression and/or amplification 11 (10%) Preoperative chemotherapy T/FAC* 66 (62%) FAC 38 (36%) Trastuzumab-containing regimen 2 (2%) Pathologic complete remission No 83 (78%) Yes 20 (19%) Not assessable 3 (3%) *: Sequential Paclitaxel / FAC (or FEC) or docetaxel /FEC (n=2) Supplementary Table 2. Minimum Common region (MCR) gained in more than 20% of samples: Band1 Probes Freq Genes AP2M1 CLCN2 DVL3 EIF4G1 EPHB3 POLR2H PSMD2 THPO -
WO 2008/104608 Al
(12) INTERNATIONAL APPLICATION PUBLISHED UNDER THE PATENT COOPERATION TREATY (PCT) (19) World Intellectual Property Organization International Bureau (43) International Publication Date PCT (10) International Publication Number 4 September 2008 (04.09.2008) WO 2008/104608 Al (51) International Patent Classification: (74) Agent: BRANTS, Johan Philippe EmUe; E. Gevaertdreef C12Q 1/68 (2006.01) 10a, B-9830 Sint-Martens-Latem (BE). (21) International Application Number: (81) Designated States (unless otherwise indicated, for every PCT/EP2008/052532 kind of national protection available): AE, AG, AL, AM, AO, AT,AU, AZ, BA, BB, BG, BH, BR, BW, BY, BZ, CA, (22) International Filing Date: CH, CN, CO, CR, CU, CZ, DE, DK, DM, DO, DZ, EC, EE, 29 February 2008 (29.02.2008) EG, ES, FI, GB, GD, GE, GH, GM, GT, HN, HR, HU, ID, IL, IN, IS, JP, KE, KG, KM, KN, KP, KR, KZ, LA, LC, (25) Filing Language: English LK, LR, LS, LT, LU, LY, MA, MD, ME, MG, MK, MN, MW, MX, MY, MZ, NA, NG, NI, NO, NZ, OM, PG, PH, (26) Publication Language: English PL, PT, RO, RS, RU, SC, SD, SE, SG, SK, SL, SM, SV, (30) Priority Data: SY, TJ, TM, TN, TR, TT, TZ, UA, UG, US, UZ, VC, VN, ZA, ZM, ZW 07447014.7 1 March 2007 (0 1.03.2007) EP (71) Applicants (for all designated States except US): UNI- (84) Designated States (unless otherwise indicated, for every VERSITE CATHOLIQUE DE LOUVAIN [BE/BE]; kind of regional protection available): ARIPO (BW, GH, Place de lUniversite 1, B-1348 Louvain-la-Neuve GM, KE, LS, MW, MZ, NA, SD, SL, SZ, TZ, UG, ZM, (BE).