Large-Scale Detection of Ubiquitination Substrates Using Cell Extracts and Protein Microarrays
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

1 ICR-Geneset Gene List
ICR-geneset Gene List. IMAGE ID UniGene Locus Name Cluster 20115 Hs.62185 SLC9A6 solute carrier family 9 (sodium/hydrogen exchanger), isoform 6 21738 21899 Hs.78353 SRPK2 SFRS protein kinase 2 21908 Hs.79133 CDH8 cadherin 8, type 2 22040 Hs.151738 MMP9 matrix metalloproteinase 9 (gelatinase B, 92kD gelatinase, 92kD type IV collagenase) 22411 Hs.183 FY Duffy blood group 22731 Hs.1787 PHRET1 PH domain containing protein in retina 1 22859 Hs.356487 ESTs 22883 Hs.150926 FPGT fucose-1-phosphate guanylyltransferase 22918 Hs.346868 EBNA1BP2 EBNA1 binding protein 2 23012 Hs.158205 BLZF1 basic leucine zipper nuclear factor 1 (JEM-1) 23073 Hs.284244 FGF2 fibroblast growth factor 2 (basic) 23173 Hs.151051 MAPK10 mitogen-activated protein kinase 10 23185 Hs.289114 TNC tenascin C (hexabrachion) 23282 Hs.8024 IK IK cytokine, down-regulator of HLA II 23353 23431 Hs.50421 RB1CC1 RB1-inducible coiled-coil 1 23514 23548 Hs.71848 Human clone 23548 mRNA sequence 23629 Hs.135587 Human clone 23629 mRNA sequence 23658 Hs.265855 SETMAR SET domain and mariner transposase fusion gene 23676 Hs.100841 Homo sapiens clone 23676 mRNA sequence 23772 Hs.78788 LZTR1 leucine-zipper-like transcriptional regulator, 1 23776 Hs.75438 QDPR quinoid dihydropteridine reductase 23804 Hs.343586 ZFP36 zinc finger protein 36, C3H type, homolog (mouse) 23831 Hs.155247 ALDOC aldolase C, fructose-bisphosphate 23878 Hs.99902 OPCML opioid binding protein/cell adhesion molecule-like 23903 Hs.12526 Homo sapiens clone 23903 mRNA sequence 23932 Hs.368063 Human clone 23932 mRNA sequence 24004 -

Download The
PROBING THE INTERACTION OF ASPERGILLUS FUMIGATUS CONIDIA AND HUMAN AIRWAY EPITHELIAL CELLS BY TRANSCRIPTIONAL PROFILING IN BOTH SPECIES by POL GOMEZ B.Sc., The University of British Columbia, 2002 A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF MASTER OF SCIENCE in THE FACULTY OF GRADUATE STUDIES (Experimental Medicine) THE UNIVERSITY OF BRITISH COLUMBIA (Vancouver) January 2010 © Pol Gomez, 2010 ABSTRACT The cells of the airway epithelium play critical roles in host defense to inhaled irritants, and in asthma pathogenesis. These cells are constantly exposed to environmental factors, including the conidia of the ubiquitous mould Aspergillus fumigatus, which are small enough to reach the alveoli. A. fumigatus is associated with a spectrum of diseases ranging from asthma and allergic bronchopulmonary aspergillosis to aspergilloma and invasive aspergillosis. Airway epithelial cells have been shown to internalize A. fumigatus conidia in vitro, but the implications of this process for pathogenesis remain unclear. We have developed a cell culture model for this interaction using the human bronchial epithelium cell line 16HBE and a transgenic A. fumigatus strain expressing green fluorescent protein (GFP). Immunofluorescent staining and nystatin protection assays indicated that cells internalized upwards of 50% of bound conidia. Using fluorescence-activated cell sorting (FACS), cells directly interacting with conidia and cells not associated with any conidia were sorted into separate samples, with an overall accuracy of 75%. Genome-wide transcriptional profiling using microarrays revealed significant responses of 16HBE cells and conidia to each other. Significant changes in gene expression were identified between cells and conidia incubated alone versus together, as well as between GFP positive and negative sorted cells. -

Predicting Clinical Response to Treatment with a Soluble Tnf-Antagonist Or Tnf, Or a Tnf Receptor Agonist
(19) TZZ _ __T (11) EP 2 192 197 A1 (12) EUROPEAN PATENT APPLICATION (43) Date of publication: (51) Int Cl.: 02.06.2010 Bulletin 2010/22 C12Q 1/68 (2006.01) (21) Application number: 08170119.5 (22) Date of filing: 27.11.2008 (84) Designated Contracting States: (72) Inventor: The designation of the inventor has not AT BE BG CH CY CZ DE DK EE ES FI FR GB GR yet been filed HR HU IE IS IT LI LT LU LV MC MT NL NO PL PT RO SE SI SK TR (74) Representative: Habets, Winand Designated Extension States: Life Science Patents AL BA MK RS PO Box 5096 6130 PB Sittard (NL) (71) Applicant: Vereniging voor Christelijk Hoger Onderwijs, Wetenschappelijk Onderzoek en Patiëntenzorg 1081 HV Amsterdam (NL) (54) Predicting clinical response to treatment with a soluble tnf-antagonist or tnf, or a tnf receptor agonist (57) The invention relates to methods for predicting a clinical response to a therapy with a soluble TNF antagonist, TNF or a TNF receptor agonist and a kit for use in said methods. EP 2 192 197 A1 Printed by Jouve, 75001 PARIS (FR) EP 2 192 197 A1 Description [0001] The invention relates to methods for predicting a clinical response to a treatment with a soluble TNF antagonist, with TNF or a TNF receptor agonist using expression levels of genes of the Type I INF pathway and a kit for use in said 5 methods. In another aspect, the invention relates to a method for evaluating a pharmacological effect of a treatment with a soluble TNF antagonist, TNF or a TNF receptor agonist. -

Association of Gene Ontology Categories with Decay Rate for Hepg2 Experiments These Tables Show Details for All Gene Ontology Categories
Supplementary Table 1: Association of Gene Ontology Categories with Decay Rate for HepG2 Experiments These tables show details for all Gene Ontology categories. Inferences for manual classification scheme shown at the bottom. Those categories used in Figure 1A are highlighted in bold. Standard Deviations are shown in parentheses. P-values less than 1E-20 are indicated with a "0". Rate r (hour^-1) Half-life < 2hr. Decay % GO Number Category Name Probe Sets Group Non-Group Distribution p-value In-Group Non-Group Representation p-value GO:0006350 transcription 1523 0.221 (0.009) 0.127 (0.002) FASTER 0 13.1 (0.4) 4.5 (0.1) OVER 0 GO:0006351 transcription, DNA-dependent 1498 0.220 (0.009) 0.127 (0.002) FASTER 0 13.0 (0.4) 4.5 (0.1) OVER 0 GO:0006355 regulation of transcription, DNA-dependent 1163 0.230 (0.011) 0.128 (0.002) FASTER 5.00E-21 14.2 (0.5) 4.6 (0.1) OVER 0 GO:0006366 transcription from Pol II promoter 845 0.225 (0.012) 0.130 (0.002) FASTER 1.88E-14 13.0 (0.5) 4.8 (0.1) OVER 0 GO:0006139 nucleobase, nucleoside, nucleotide and nucleic acid metabolism3004 0.173 (0.006) 0.127 (0.002) FASTER 1.28E-12 8.4 (0.2) 4.5 (0.1) OVER 0 GO:0006357 regulation of transcription from Pol II promoter 487 0.231 (0.016) 0.132 (0.002) FASTER 6.05E-10 13.5 (0.6) 4.9 (0.1) OVER 0 GO:0008283 cell proliferation 625 0.189 (0.014) 0.132 (0.002) FASTER 1.95E-05 10.1 (0.6) 5.0 (0.1) OVER 1.50E-20 GO:0006513 monoubiquitination 36 0.305 (0.049) 0.134 (0.002) FASTER 2.69E-04 25.4 (4.4) 5.1 (0.1) OVER 2.04E-06 GO:0007050 cell cycle arrest 57 0.311 (0.054) 0.133 (0.002) -

Dissertation
Regulation of gene silencing: From microRNA biogenesis to post-translational modifications of TNRC6 complexes DISSERTATION zur Erlangung des DOKTORGRADES DER NATURWISSENSCHAFTEN (Dr. rer. nat.) der Fakultät Biologie und Vorklinische Medizin der Universität Regensburg vorgelegt von Johannes Danner aus Eggenfelden im Jahr 2017 Das Promotionsgesuch wurde eingereicht am: 12.09.2017 Die Arbeit wurde angeleitet von: Prof. Dr. Gunter Meister Johannes Danner Summary ‘From microRNA biogenesis to post-translational modifications of TNRC6 complexes’ summarizes the two main projects, beginning with the influence of specific RNA binding proteins on miRNA biogenesis processes. The fate of the mature miRNA is determined by the incorporation into Argonaute proteins followed by a complex formation with TNRC6 proteins as core molecules of gene silencing complexes. miRNAs are transcribed as stem-loop structured primary transcripts (pri-miRNA) by Pol II. The further nuclear processing is carried out by the microprocessor complex containing the RNase III enzyme Drosha, which cleaves the pri-miRNA to precursor-miRNA (pre-miRNA). After Exportin-5 mediated transport of the pre-miRNA to the cytoplasm, the RNase III enzyme Dicer cleaves off the terminal loop resulting in a 21-24 nt long double-stranded RNA. One of the strands is incorporated in the RNA-induced silencing complex (RISC), where it directly interacts with a member of the Argonaute protein family. The miRNA guides the mature RISC complex to partially complementary target sites on mRNAs leading to gene silencing. During this process TNRC6 proteins interact with Argonaute and recruit additional factors to mediate translational repression and target mRNA destabilization through deadenylation and decapping leading to mRNA decay. -

Nercc1, a Mammalian NIMA-Family Kinase, Binds the Ran Gtpase and Regulates Mitotic Progression
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Nercc1, a mammalian NIMA-family kinase, binds the Ran GTPase and regulates mitotic progression Joan Roig, Alexei Mikhailov, Christopher Belham, and Joseph Avruch1 Department of Molecular Biology and the Diabetes Unit and Medical Services, Massachusetts General Hospital, and the Department of Medicine, Harvard Medical School, Boston, Massachusetts 02114, USA The protein kinase NIMA is an indispensable pleiotropic regulator of mitotic progression in Aspergillus. Although several mammalian NIMA-like kinases (Neks) are known, none appears to have the broad importance for mitotic regulation attributed to NIMA. Nercc1 is a new NIMA-like kinase that regulates chromosome alignment and segregation in mitosis. Its NIMA-like catalytic domain is followed by a noncatalytic tail containing seven repeats homologous to those of the Ran GEF, RCC1, a Ser/Thr/Pro-rich segment, and a coiled-coil domain. Nercc1 binds to another NIMA-like kinase, Nek6, and also binds specifically to the Ran GTPase through both its catalytic and its RCC1-like domains, preferring RanGDP in vivo. Nercc1 exists as a homooligomer and can autoactivate in vitro by autophosphorylation. Nercc1 is a cytoplasmic protein that is activated during mitosis and is avidly phosphorylated by active p34Cdc2. Microinjection of anti-Nercc1 antibodies in prophase results in spindle abnormalities and/or chromosomal misalignment. In Ptk2 cells the outcome is prometaphase arrest or aberrant chromosome segregation and aneuploidy, whereas in CFPAC-1 cells prolonged arrest in prometaphase is the usual response. Nercc1 and its partner Nek6 represent a new signaling pathway that regulates mitotic progression. -

Characterisation of Isomirs in Stem Cells
Characterisation of isomiRs in stem cells Geok Chin Tan Institute of Reproductive and Developmental Biology Department of Surgery and Cancer Faculty of Medicine Imperial College London Thesis submitted to Imperial College London for the degree of Doctor of Philosophy 1 Statement of Originality All experiments included in this thesis were performed by me unless otherwise stated in the text. 2 Copyright Statement ‘The copyright of this thesis rests with the author and is made available under a Creative Commons Attribution Non-Commercial No Derivatives licence. Researchers are free to copy, distribute or transmit the thesis on the condition that they attribute it, that they do not use it for commercial purposes and that they do not alter, transform or build upon it. For any reuse or redistribution, researchers must make clear to others the licence terms of this work’ 3 Acknowledgements I would like to thank my supervisor Dr Nicholas Dibb for giving me the opportunity to work in his lab and for all of his guidance and support throughout my PhD, without which this project would not have been possible. I am also very grateful to Dr Wei Cui for teaching me the technique of stem cell culture, her comments on my project related to stem cells and as a wonderful co-supervisor. I would like to also thank Professor Malcolm Parker for his supports and advise on academic and non-academic related subjects. Many thanks to Elcie Chan for the generation of all the stem cell libraries which forms the platform for my project. My sincere thanks also to Gunter Meister for supplying the Argonaute antibodies, Leandro Castellano for the help in the design of RNA sponges, Laki Buluwela for the pTRIPz lentiviral vector and last but not least Alywn Dart from Charlotte Bevan group for the prostate cancer cell lines. -
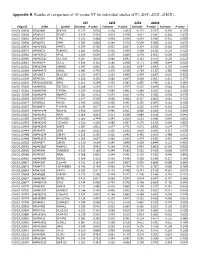
Appendix B. Results of Comparison of AI Versus NT for Individual Studies (D7e, D25e, D25x, D280x)
Appendix B. Results of comparison of AI versus NT for individual studies (d7E, d25E, d25X, d280X). d7E d25E d25X d280X Oligo ID cDNA Symbol Estimate P-value Estimate P-value Estimate P-value Estimate P-value OLIGO_00038 BF042860 RNF166 -0.123 0.634 0.194 0.469 -0.171 0.537 -0.302 0.263 OLIGO_00040 BF045167 POMT1 0.019 0.903 0.010 0.978 0.411 0.181 -0.204 0.476 OLIGO_00043 BF043567 ETFA 0.131 0.484 -0.094 0.706 -0.053 0.745 -0.331 0.056 OLIGO_00068 BF042951 GPX3 0.023 0.840 -0.249 0.370 -0.290 0.088 0.181 0.437 OLIGO_00070 AW464501 HPRT1 -0.079 0.785 0.055 0.837 0.224 0.259 -0.220 0.097 OLIGO_00074 BF040652 PLEKHC1 0.085 0.691 -0.256 0.399 0.309 0.156 0.133 0.423 OLIGO_00081 AW463422 PPIL1 0.109 0.600 0.142 0.686 0.379 0.061 -0.609 0.003 OLIGO_00086 AW462335 SULT1A1 -0.027 0.870 -0.083 0.845 0.123 0.675 0.520 0.121 OLIGO_00087 BF046074 CCL15 -0.306 0.132 -0.585 0.038 -0.771 0.038 0.844 0.020 OLIGO_00161 BM362588 USMG5 0.047 0.836 -0.136 0.628 0.547 0.043 0.279 0.064 OLIGO_00166 AW464473 UFD1L 0.083 0.742 0.372 0.538 0.759 0.132 0.285 0.418 OLIGO_00196 BF039617 BLOC1S2 0.011 0.972 0.153 0.699 0.424 0.187 0.030 0.931 OLIGO_00209 BF045356 GRB2 0.226 0.156 0.685 0.007 -0.008 0.961 0.212 0.177 OLIGO_00220 BP230002B20E6 NFX1 -0.161 0.294 -0.133 0.583 0.055 0.746 -0.211 0.290 OLIGO_00228 AW465641 TBC1D12 -0.248 0.236 -0.013 0.975 -0.223 0.496 0.044 0.821 OLIGO_00261 AW463949 TATDN1 0.020 0.914 -0.089 0.861 -0.248 0.611 -0.412 0.264 OLIGO_00308 AW464976 SNAPC3 0.021 0.911 -0.233 0.417 -0.054 0.782 0.004 0.979 OLIGO_00329 BF045997 PTP4A1 0.247 0.250 -

A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita
G C A T T A C G G C A T genes Article A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita Xenia Latypova 1, Stefan Giovanni Creadore 2, Noémi Dahan-Oliel 3,4, Anxhela Gjyshi Gustafson 2, Steven Wei-Hung Hwang 5, Tanya Bedard 6, Kamran Shazand 2, Harold J. P. van Bosse 5 , Philip F. Giampietro 7,* and Klaus Dieterich 8,* 1 Grenoble Institut Neurosciences, Université Grenoble Alpes, Inserm, U1216, CHU Grenoble Alpes, 38000 Grenoble, France; [email protected] 2 Shriners Hospitals for Children Headquarters, Tampa, FL 33607, USA; [email protected] (S.G.C.); [email protected] (A.G.G.); [email protected] (K.S.) 3 Shriners Hospitals for Children, Montreal, QC H4A 0A9, Canada; [email protected] 4 School of Physical & Occupational Therapy, Faculty of Medicine and Health Sciences, McGill University, Montreal, QC H3G 2M1, Canada 5 Shriners Hospitals for Children, Philadelphia, PA 19140, USA; [email protected] (S.W.-H.H.); [email protected] (H.J.P.v.B.) 6 Alberta Congenital Anomalies Surveillance System, Alberta Health Services, Edmonton, AB T5J 3E4, Canada; [email protected] 7 Department of Pediatrics, University of Illinois-Chicago, Chicago, IL 60607, USA 8 Institut of Advanced Biosciences, Université Grenoble Alpes, Inserm, U1209, CHU Grenoble Alpes, 38000 Grenoble, France * Correspondence: [email protected] (P.F.G.); [email protected] (K.D.) Citation: Latypova, X.; Creadore, S.G.; Dahan-Oliel, N.; Gustafson, Abstract: Arthrogryposis multiplex congenita (AMC) describes a group of conditions characterized A.G.; Wei-Hung Hwang, S.; Bedard, by the presence of non-progressive congenital contractures in multiple body areas. -

A SARS-Cov-2 Protein Interaction Map Reveals Targets for Drug Repurposing
Article A SARS-CoV-2 protein interaction map reveals targets for drug repurposing https://doi.org/10.1038/s41586-020-2286-9 A list of authors and affiliations appears at the end of the paper Received: 23 March 2020 Accepted: 22 April 2020 A newly described coronavirus named severe acute respiratory syndrome Published online: 30 April 2020 coronavirus 2 (SARS-CoV-2), which is the causative agent of coronavirus disease 2019 (COVID-19), has infected over 2.3 million people, led to the death of more than Check for updates 160,000 individuals and caused worldwide social and economic disruption1,2. There are no antiviral drugs with proven clinical efcacy for the treatment of COVID-19, nor are there any vaccines that prevent infection with SARS-CoV-2, and eforts to develop drugs and vaccines are hampered by the limited knowledge of the molecular details of how SARS-CoV-2 infects cells. Here we cloned, tagged and expressed 26 of the 29 SARS-CoV-2 proteins in human cells and identifed the human proteins that physically associated with each of the SARS-CoV-2 proteins using afnity-purifcation mass spectrometry, identifying 332 high-confdence protein–protein interactions between SARS-CoV-2 and human proteins. Among these, we identify 66 druggable human proteins or host factors targeted by 69 compounds (of which, 29 drugs are approved by the US Food and Drug Administration, 12 are in clinical trials and 28 are preclinical compounds). We screened a subset of these in multiple viral assays and found two sets of pharmacological agents that displayed antiviral activity: inhibitors of mRNA translation and predicted regulators of the sigma-1 and sigma-2 receptors. -

Ncomms6059.Pdf
ARTICLE Received 4 Feb 2014 | Accepted 21 Aug 2014 | Published 6 Oct 2014 DOI: 10.1038/ncomms6059 OPEN ABRO1 suppresses tumourigenesis and regulates the DNA damage response by stabilizing p53 Jianhong Zhang1,*, Mengmeng Cao2,*, Jiahong Dong3, Changyan Li1, Wangxiang Xu1, Yiqun Zhan1, Xiaohui Wang1, Miao Yu1, Changhui Ge1, Zhiqiang Ge2 & Xiaoming Yang1,2 Abraxas brother 1 (ABRO1) has been reported to be a component of the BRISC complex, a multiprotein complex that specifically cleaves ‘Lys-63’-linked ubiquitin. However, current knowledge of the functions of ABRO1 is limited. Here we report that ABRO1 is frequently downregulated in human liver, kidney, breast and thyroid gland tumour tissues. Depletion of ABRO1 in cancer cells reduces p53 levels and enhances clone formation and cellular transformation. Conversely, overexpression of ABRO1 suppresses cell proliferation and tumour formation in a p53- dependent manner. We further show that ABRO1 stabilizes p53 by facilitating the interaction of p53 with USP7. DNA-damage induced accumulation of endogenous ABRO1 as well as translocation of ABRO1 to the nucleus, and the induction of p53 by DNA damage is almost completely attenuated by ABRO1 depletion. Our study shows that ABRO1 is a novel p53 regulator that plays an important role in tumour suppression and the DNA damage response. 1 State Key Laboratory of Proteomics, Beijing Proteomics Research Center, Beijing Institute of Radiation Medicine, 27-Taiping Road, Beijing 100850, China. 2 School of Chemical Engineering and Technology, TianJin University, Tianjin 300073, China. 3 Department of Liver and Gallbladder Surgery, The General Hospital of Chinese People’s Liberation Army, Beijing 100853, China. * These authors contributed equally to this work. -

Page 1 Exploring the Understudied Human Kinome For
bioRxiv preprint doi: https://doi.org/10.1101/2020.04.02.022277; this version posted June 30, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Exploring the understudied human kinome for research and therapeutic opportunities Nienke Moret1,2,*, Changchang Liu1,2,*, Benjamin M. Gyori2, John A. Bachman,2, Albert Steppi2, Rahil Taujale3, Liang-Chin Huang3, Clemens Hug2, Matt Berginski1,4,5, Shawn Gomez1,4,5, Natarajan Kannan,1,3 and Peter K. Sorger1,2,† *These authors contributed equally † Corresponding author 1The NIH Understudied Kinome Consortium 2Laboratory of Systems Pharmacology, Department of Systems Biology, Harvard Program in Therapeutic Science, Harvard Medical School, Boston, Massachusetts 02115, USA 3 Institute of Bioinformatics, University of Georgia, Athens, GA, 30602 USA 4 Department of Pharmacology, The University of North Carolina at Chapel Hill, Chapel Hill, NC 27599, USA 5 Joint Department of Biomedical Engineering at the University of North Carolina at Chapel Hill and North Carolina State University, Chapel Hill, NC 27599, USA Key Words: kinase, human kinome, kinase inhibitors, drug discovery, cancer, cheminformatics, † Peter Sorger Warren Alpert 432 200 Longwood Avenue Harvard Medical School, Boston MA 02115 [email protected] cc: [email protected] 617-432-6901 ORCID Numbers Peter K. Sorger 0000-0002-3364-1838 Nienke Moret 0000-0001-6038-6863 Changchang Liu 0000-0003-4594-4577 Ben Gyori 0000-0001-9439-5346 John Bachman 0000-0001-6095-2466 Albert Steppi 0000-0001-5871-6245 Page 1 bioRxiv preprint doi: https://doi.org/10.1101/2020.04.02.022277; this version posted June 30, 2020.