Convergent Functional Genomics: a Bayesian Candidate Gene Identiwcation Approach for Complex Disorders
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Runx1 Gene in Acute Myeloid Leukemia: Expression Level Signifcance and Impact on Clinical Features
Runx1 Gene in Acute Myeloid Leukemia: Expression Level Signicance and Impact on Clinical Features Fadwa Said Abdelazim Mohamed ( [email protected] ) Cairo University Roxan E Shak National Cancer Institute, Cairo University Naglaa M. Hassan National Cancer Institute, Cairo University Research article Keywords: Acute myeloid leukemia, RUNX1, mutation, gene expression, Egypt Posted Date: April 7th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-20926/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/20 Abstract Background Acute myeloid leukemia represents the highest percentage of all adult acute leukemia variants. Runt-related transcription factor1 (RUNX1), a transcription factor with a known tumor suppressor function was recently reported as a tumor promotor in AML. We investigated the role of RUNX1 geneexpression levelin Egyptian AML patients and delineateits clinical signicance. Methods This study recruited 91 AML patients that were recently diagnosed at our hospital with 14 healthy age- and sex-matched donors of bone marrow transplantation unit. We measured RUNX1 gene expression level using reverse transcription–quantitative polymerase chain reaction. Results We found that RUNX1 gene expression level was signicantly higher compared to the control group (p < 0.001). Patients with FLT3 mutations had the higher expression level of RUNX1 (p=0.023). The male patients expressed signicantly higher level of RUNX1 (p=0.046). Conclusion The RUNX1 gene expression may serve as a diagnostic marker in Egyptian AML patients. Its relation to FLT3 may give clue that patients carrying this mutation may benet fromnew treatments that target RUNX1 in the future. -
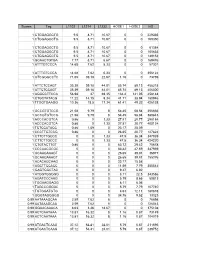
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 229335 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 169350 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 61384 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 105633 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 149152 1 GCAACTGTGA 7.77 8.71 6.67 0 0 169476 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 57301 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 356122 1 GTCGGGCCTC 71.65 39.18 22.67 1.16 0 73769 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 458218 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 406300 1 AGGGCTTCCA 56.98 37 69.35 134.4 141.05 458148 1 CTGCTATACG 11.22 14.15 9.34 41.71 38.94 180946 1 TTGGTGAAGG 10.36 18.5 17.34 61.41 49.32 426138 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 356666 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 380843 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 298184 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 400124 1 TCTCCATACC 0.86 1.09 0 23.17 25.09 1 CCCTTGTCCG 0.86 0 0 26.65 20.77 127824 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 347939 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 424220 1 CTGTACTTGT 0.86 0 0 63.72 29.42 75678 1 CCCAACGCGC 0 0 0 83.42 47.59 347939 1 GCAAGAAAGT 0 0 0 26.65 39.81 36977 1 GCAAGAAAGT 0 0 0 26.65 39.81 155376 1 ACACAGCAAG 0 0 0 23.17 15.58 1 AGCTTCCACC 0 0 0 11.59 7.79 355542 1 GAGTGGCTAC 0 0 0 9.27 6.92 1 ATGGTGGGGG 0 0 0 8.11 22.5 343586 1 AGATCCCAAG 0 0 0 5.79 8.65 50813 1 TGGAAGGAGG 0 0 0 8.11 6.06 1 TAGCCGGGAC 0 0 0 5.79 7.79 107740 1 TGTGGATGTG 0 0 0 4.63 12.11 180878 1 GGGTAGGGGG 0 0 0 34.76 9.52 13323 0.99 AATAAAGCAA 2.59 7.62 8 0 0 76698 0.99 AATAAAGCAA 2.59 7.62 8 0 0 126043 0.99 GGAACAAACA 8.63 3.26 18.67 0 0 375108 -

(12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer Et Al
USOO8440393B2 (12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer et al. (45) Date of Patent: May 14, 2013 (54) PRO-ANGIOGENIC GENES IN OVARIAN OTHER PUBLICATIONS TUMORENDOTHELIAL CELL, SOLATES Boyd (The Basic Science of Oncology, 1992, McGraw-Hill, Inc., p. (75) Inventors: Michael J. Birrer, Mt. Airy, MD (US); 379). Tomas A. Bonome, Washington, DC Tockman et al. (Cancer Res., 1992, 52:2711s-2718s).* (US); Anil Sood, Pearland, TX (US); Pritzker (Clinical Chemistry, 2002, 48: 1147-1150).* Chunhua Lu, Missouri City, TX (US) Benedict et al. (J. Exp. Medicine, 2001, 193(1) 89-99).* Jiang et al. (J. Biol. Chem., 2003, 278(7) 4763-4769).* (73) Assignees: The United States of America as Matsushita et al. (FEBS Letters, 1999, vol. 443, pp. 348-352).* Represented by the Secretary of the Singh et al. (Glycobiology, 2001, vol. 11, pp. 587-592).* Department of Health and Human Abbosh et al. (Cancer Res. Jun. 1, 2006 66:5582-55.91 and Supple Services, Washington, DC (US); The mental Figs. S1-S7).* University of MD Anderson Cancer Zhai et al. (Chinese General Practice Aug. 2008, 11(8A): 1366 Center, Houston, TX (US) 1367).* Lu et al. (Cancer Res. Feb. 15, 2007, 64(4): 1757-1768).* (*) Notice: Subject to any disclaimer, the term of this Bagnato et al., “Activation of Mitogenic Signaling by Endothelin 1 in patent is extended or adjusted under 35 Ovarian Carcinoma Cells', Cancer Research, vol. 57, pp. 1306-1311, U.S.C. 154(b) by 194 days. 1997. Bouras et al., “Stanniocalcin 2 is an Estrogen-responsive Gene (21) Appl. -

An Optimized Streptavidin-Binding RNA Aptamer for Purification Of
Published online 23 October 2013 Nucleic Acids Research, 2014, Vol. 42, No. 2 e13 doi:10.1093/nar/gkt956 An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins Kathrin Leppek1,2,3 and Georg Stoecklin1,2,3,* 1Helmholtz Junior Research Group Posttranscriptional Control of Gene Expression, German Cancer Research Center (DKFZ), Im Neuenheimer Feld 280, 69120 Heidelberg, Germany, 2Zentrum fu¨ r Molekulare Biologie der Universita¨ t Heidelberg (ZMBH), Im Neuenheimer Feld 282, 69120 Heidelberg, Germany and 3DKFZ-ZMBH Downloaded from https://academic.oup.com/nar/article-abstract/42/2/e13/1030103 by guest on 11 March 2019 Alliance Received June 19, 2013; Revised September 6, 2013; Accepted September 27, 2013 ABSTRACT degradation (1,2). The fate of any given mRNA is essen- tially determined by the messenger ribonucleoprotein Determining the composition of messenger (mRNP) complex, i.e. the ensemble of proteins and regu- ribonucleoprotein (mRNP) particles is essential for latory RNAs an mRNA interacts with. Although decades a comprehensive understanding of the complex of research have uncovered specific functions of numerous mechanisms underlying mRNA regulation, but is RNA-binding proteins (RNA-BPs), surprisingly little is technically challenging. Here we present an RNA- known about the composition, heterogeneity and based method to identify RNP components using a dynamics of whole mRNPs. modified streptavidin (SA)-binding RNA aptamer There are essentially two strategies to explore the termed S1m. By optimizing the RNA aptamer S1 in content of mRNPs. On one hand, protein-based structure and repeat conformation, we improved its approaches rely on the purification of a particular RNA- affinity for SA and found a 4-fold repeat of S1m BP by immunoprecipitation together with its associated mRNAs, whose identity can then be determined by (4ÂS1m) to be more efficient than the established cDNA cloning, microarray analysis (RIP-Chip) or deep MS2 and PP7 systems from bacteriophages. -

Candidate Genes, Pathways and Mechanisms for Bipolar (Manic–Depressive) and Related Disorders: an Expanded Convergent Function
Molecular Psychiatry (2004), 1007–1029 & 2004 Nature Publishing Group All rights reserved 1359-4184/04 $30.00 www.nature.com/mp ORIGINAL RESEARCH ARTICLE Candidate genes, pathways and mechanisms for bipolar (manic–depressive) and related disorders: an expanded convergent functional genomics approach CA Ogden1,2,3, ME Rich1,2,3, NJ Schork2, MP Paulus2,3, MA Geyer2,3, JB Lohr2,3, R Kuczenski2,3 and AB Niculescu1,2,3 1Laboratory of Neurophenomics, University of California, San Diego, CA, USA; 2Department of Psychiatry, University of California, San Diego, CA, USA; 3VA San Diego Healthcare System VISN-22 MIRECC, San Diego, CA, USA Identifying genes for bipolar mood disorders through classic genetics has proven difficult. Here, we present a comprehensive convergent approach that translationally integrates brain gene expression data from a relevant pharmacogenomic mouse model (involving treatments with a stimulant—methamphetamine, and a mood stabilizer—valproate), with human data (linkage loci from human genetic studies, changes in postmortem brains from patients), as a bayesian strategy of crossvalidating findings. Topping the list of candidate genes, we have DARPP-32 (dopamine- and cAMP-regulated phosphoprotein of 32 kDa) located at 17q12, PENK (preproenkephalin) located at 8q12.1, and TAC1 (tachykinin 1, substance P) located at 7q21.3. These data suggest that more primitive molecular mechanisms involved in pleasure and pain may have been recruited by evolution to play a role in higher mental functions such as mood. The analysis also revealed other high-probability candidates genes (neurogenesis, neuro- trophic, neurotransmitter, signal transduction, circadian, synaptic, and myelin related), pathways and mechanisms of likely importance in pathophysiology. -

(12) Patent Application Publication (10) Pub. No.: US 2013/0274315 A1 Birrer Et Al
US 201302743 15A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0274315 A1 Birrer et al. (43) Pub. Date: Oct. 17, 2013 (54) PRO-ANGIOGENC GENES IN OVARAN (60) Provisional application No. 60/901,455, filed on Feb. TUMORENDOTHELIAL CELL, SOLATES 14, 2007. (71) Applicants: The University of Texas MD Anderson Publication Classification Cancer Center, Houston, TX (US); The Government of the U.S.A as (51) Int. Cl. represented by the Secretary of the CI2O I/68 (2006.01) Department of He, Rockville, MD (US) (52) U.S. Cl. CPC .................................... CI2O I/6886 (2013.01) (72) Inventors: Michael J. Birrer, Mt. Airy, MD (US); USPC ............... 514/44A: 435/6.12: 506/9: 435/7.1 Tomas A. Bonome, Washington, DC (US); Anil Sood, Pearland, TX (US); (57) ABSTRACT Chunhua Lu, Missouri City, TX (US) A gene profiling signature for ovarian tumor endothelial cells is disclosed herein. The gene signature can be used to diag nosis or prognosis an ovarian tumor, identify agents to treat an (21) Appl. No.: 13/863,219 ovariantumor, to predict the metastatic potential of an ovarian tumor and to determine the effectiveness of ovarian tumor (22) Filed: Apr. 15, 2013 treatments. Thus, methods are provided for identifying agents that can be used to treat ovarian cancer, for determining the effectiveness of an ovarian tumor treatment, or to diagnose or Related U.S. Application Data prognose an ovarian tumor. Methods of treatment are also (60) Division of application No. 12/541,729, filed on Aug. disclosed which include administering a composition that 14, 2009, now Pat. -

Characterization of Molecular Targets for Differential Regulation of the Type I and III Interferon Induction and Signalling Pathways by Rotavirus NSP1
Characterization of molecular targets for differential regulation of the type I and III interferon induction and signalling pathways by rotavirus NSP1 Thesis submitted in accordance with the requirements of the University of Liverpool for the degree of Doctor in Philosophy by Gennaro Iaconis September 2018 Table of contents TABLE OF CONTENTS ............................................................................................................................... 1 LIST OF FIGURES ...................................................................................................................................... 6 LIST OF TABLES ........................................................................................................................................ 8 DECLARATION ....................................................................................................................................... 10 ABSTRACT ............................................................................................................................................. 11 1 INTRODUCTION ........................................................................................................................... 12 1.1 Rotavirus ................................................................................................................. 12 1.1.1 Historical Background ......................................................................................... 12 1.1.2 Classification ...................................................................................................... -

The Induction Levels of Heat Shock Protein 70 Differentiate the Vulnerabilities to Mutant Huntingtin Among Neuronal Subtypes
868 • The Journal of Neuroscience, January 24, 2007 • 27(4):868–880 Neurobiology of Disease The Induction Levels of Heat Shock Protein 70 Differentiate the Vulnerabilities to Mutant Huntingtin among Neuronal Subtypes Kazuhiko Tagawa,1 Shigeki Marubuchi,1,2 Mei-Ling Qi,1,3 Yasushi Enokido,1 Takuya Tamura,1 Reina Inagaki,1 Miho Murata3 Ichiro Kanazawa,4 Erich E. Wanker,5 and Hitoshi Okazawa1,3,6 1Department of Neuropathology, Medical Research Institute and 21st Century Center of Excellence Program for Brain Integration and Its Disorders, Tokyo Medical and Dental University, Tokyo 113-8510, Japan, 2Toyama Chemical Company, Toyama 930-8508, Japan, 3PRESTO, Japan Science and Technology Agency, Kawagoe 332-0012, Japan, 4National Center for Neurology and Psychiatry, Kodaira 187-8502, Japan, and 5Max-Delbru¨ck Center for Molecular Medicine, D-13125 Berlin, Germany The reason why vulnerabilities to mutant polyglutamine (polyQ) proteins are different among neuronal subtypes is mostly unknown. In this study, we compared the gene expression profiles of three types of primary neurons expressing huntingtin (htt) or ataxin-1. We found that heat shock protein 70 (hsp70), a well known chaperone molecule protecting neurons in the polyQ pathology, was dramatically upregulated only by mutant htt and selectively in the granule cells of the cerebellum. Granule cells, which are insensitive to degeneration in the human Huntington’s disease (HD) pathology, lost their resistance by suppressing hsp70 with siRNA, whereas cortical neurons, affected in human HD, gained resistance by overexpressing hsp70. This indicates that induction levels of hsp70 are a critical factor for determining vulnerabilities to mutant htt among neuronal subtypes. -

Roxan Sirna (H): Sc-76427
SANTA CRUZ BIOTECHNOLOGY, INC. RoXaN siRNA (h): sc-76427 BACKGROUND STORAGE AND RESUSPENSION RoXaN (zinc finger CCCH-type containing 7B), is a 993 amino acid protein Store lyophilized siRNA duplex at -20° C with desiccant. Stable for at least likely involved in translational regulation whose alternative names include one year from the date of shipment. Once resuspended, store at -20° C, rotavirus “X”-associated non-structural protein, ubiquitous tetratricopeptide avoid contact with RNAses and repeated freeze thaw cycles. containing protein RoXaN, ZC3H7B, FLJ13787, KIAA1031, or DKFZp434K0920. Resuspend lyophilized siRNA duplex in 330 µl of the RNAse-free water RoXaN localizes to the nucleus and has been identified as a novel cellular provided. Resuspension of the siRNA duplex in 330 µl of RNAse-free water protein-binding partner for the rotavirus nonstructural protein NSP3, which makes a 10 µM solution in a 10 µM Tris-HCl, pH 8.0, 20 mM NaCl, 1 mM is involved in rotavirus replication. RoXaN’s nuclear localization is known to EDTA buffered solution. appear disguised upon infection with rotavirus A. Two RoXaN isoforms exist as a result of alternative splicing, and RoXaN also contains one C H -type 2 2 APPLICATIONS zinc finger, three TPR repeats and four C3H1-type zinc fingers. RoXaN con- tains one paxillin leucine-aspartate repeat (LD) motif in addition to a coiled- RoXaN siRNA (h) is recommended for the inhibition of RoXaN expression in coil region which assist in protein-protein interactions. The gene encoding human cells. RoXaN maps to human chromosome 22q13.1. SUPPORT REAGENTS REFERENCES For optimal siRNA transfection efficiency, Santa Cruz Biotechnology’s 1. -

Tinkering with Translation: Protein Synthesis in Virus-Infected Cells
Downloaded from http://cshperspectives.cshlp.org/ on September 25, 2021 - Published by Cold Spring Harbor Laboratory Press Tinkering with Translation: Protein Synthesis in Virus-Infected Cells Derek Walsh1, Michael B. Mathews2, and Ian Mohr1 1Department of Microbiology, New York University School of Medicine, New York, New York 10016 2Department of Biochemistry and Molecular Biology, UMDNJ–New Jersey Medical School, University of Medicine and Dentistry New Jersey, Newark, New Jersey 07103-1709 Correspondence: [email protected]; [email protected]; [email protected] Viruses are obligate intracellular parasites, and their replication requires host cell functions. Although the size, composition, complexity, and functions encoded by their genomes are remarkably diverse, all viruses relyabsolutely on the protein synthesis machinery of their host cells. Lacking their own translational apparatus, they must recruit cellular ribosomes in order to translate viral mRNAs and produce the protein products required for their replication. In addition, there are other constraints on viral protein production. Crucially, host innate de- fenses and stress responses capable of inactivating the translation machinery must be effec- tively neutralized. Furthermore, the limited coding capacity of the viral genome needs to be used optimally.These demands have resulted in complex interactions between virus and host that exploit ostensibly virus-specific mechanisms and, at the same time, illuminate the functioning of the cellular protein synthesis apparatus. he dependence of viruses on the host trans- lation of key eukaryotic translation factors to Tlation system imposes constraints that are the evolution of specialized cis-acting elements central to virus biology and have led to special- that recruit ribosomes or modify genome-cod- ized mechanisms and intricate regulatory inter- ing capacity. -
Challenging the Roles of NSP3 and Untranslated Regions in Rotavirus Mrna Translation
Challenging the Roles of NSP3 and Untranslated Regions in Rotavirus mRNA Translation. Matthieu Gratia, Patrice Vende, Annie Charpilienne, Hilma Carolina Baron, Cécile Laroche, Emeline Sarot, Stéphane Pyronnet, Mariela Duarte, Didier Poncet To cite this version: Matthieu Gratia, Patrice Vende, Annie Charpilienne, Hilma Carolina Baron, Cécile Laroche, et al.. Challenging the Roles of NSP3 and Untranslated Regions in Rotavirus mRNA Translation.. PLoS ONE, Public Library of Science, 2015, 11 (1), pp.e0145998. 10.1371/journal.pone.0145998. hal- 01344143 HAL Id: hal-01344143 https://hal.archives-ouvertes.fr/hal-01344143 Submitted on 28 May 2020 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Distributed under a Creative Commons Attribution| 4.0 International License RESEARCH ARTICLE Challenging the Roles of NSP3 and Untranslated Regions in Rotavirus mRNA Translation Matthieu Gratia1, Patrice Vende1, Annie Charpilienne1, Hilma Carolina Baron1, Cécile Laroche1, Emeline Sarot3, Stéphane Pyronnet3, Mariela Duarte1,2, Didier Poncet1* 1 Institut de Biologie Integrative de la Cellule (I2BC), UMR 9198, Département de Virologie, USC INRA 1358, Gif sur Yvette, France, 2 Université d’Evry Val d’Essonne, Département de Biologie, Evry, France, 3 INSERM UMR-1037 - Université de Toulouse III-Paul Sabatier, Laboratoire d'Excellence Toulouse Cancer (TOUCAN), Equipe labellisée Ligue Contre le Cancer Toulouse, France a11111 * [email protected] Abstract Rotavirus NSP3 is a translational surrogate of the PABP-poly(A) complex for rotavirus OPEN ACCESS mRNAs. -

Noncoding Rnps of Viral Origin
Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Noncoding RNPs of Viral Origin Joan Steitz, Sumit Borah, Demian Cazalla, Victor Fok, Robin Lytle, Rachel Mitton-Fry, Kasandra Riley, and Tasleem Samji Department of Molecular Biophysics and Biochemistry, Howard Hughes Medical Institute, Yale University School of Medicine, New Haven, Connecticut 06536-0812 Correspondence: [email protected] SUMMARY Like their host cells, many viruses produce noncoding (nc)RNAs. These show diversity with respect to time of expression during viral infection, length and structure, protein-binding partners and relative abundance compared with their host-cell counterparts. Viruses, with their limited genomic capacity, presumably evolve or acquire ncRNAs only if they selec- tively enhance the viral life cycle or assist the virus in combating the host’s response to infection. Despite much effort, identifying the functions of viral ncRNAs has been extremely challenging. Recent technical advances and enhanced understanding of host-cell ncRNAs promise accelerated insights into the RNAwarfare mounted by this fascinating class of RNPs. Outline 1 VA RNAs: Multifunctional manipulators of 5 Viral microRNAs: Altering host-cell or adenovirus host-cell functions fine-tuning viral gene expression? 2 EBERs: Abundant but enigmatic 6 Noncoding RNAs of other flavors contributors to EBV latency 7 Prospects 3 HSURs: Managing host microRNA References functions during HVS latency 4 PAN RNA: A nuclear sink during the KSHV lytic cycle? Editors: John F. Atkins, Raymond F. Gesteland, and Thomas R. Cech Additional Perspectives on RNA Worlds available at www.cshperspectives.org Copyright # 2011 Cold Spring Harbor Laboratory Press; all rights reserved; doi: 10.1101/cshperspect.a005165 Cite this article as Cold Spring Harb Perspect Biol 2011;3:a005165 1 Downloaded from http://cshperspectives.cshlp.org/ on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press J.