An Optimized Streptavidin-Binding RNA Aptamer for Purification Of
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Supplemental Material Table of Contents
Supplemental material Table of Contents Detailed Materials and Methods ......................................................................................................... 2 Perioperative period ........................................................................................................................... 2 Ethical aspects ................................................................................................................................... 4 Evaluation of heart failure ................................................................................................................. 4 Sample preparation for ANP mRNA expression .................................................................................. 5 Sample preparation for validative qRT-PCR (Postn, Myh7, Gpx3, Tgm2) ............................................ 6 Tissue fibrosis .................................................................................................................................... 7 Ventricular remodeling and histological tissue preservation ................................................................ 8 Evaluation of the histological preservation of cardiac tissue ................................................................ 9 Sample preparation and quantitative label-free proteomics analyses .................................................. 10 Statistical methods ........................................................................................................................... 12 References ........................................................................................................................................ -

Datasheet: VPA00331KT Product Details
Datasheet: VPA00331KT Description: PRPF19 ANTIBODY WITH CONTROL LYSATE Specificity: PRPF19 Format: Purified Product Type: PrecisionAb™ Polyclonal Isotype: Polyclonal IgG Quantity: 2 Westerns Product Details Applications This product has been reported to work in the following applications. This information is derived from testing within our laboratories, peer-reviewed publications or personal communications from the originators. Please refer to references indicated for further information. For general protocol recommendations, please visit www.bio-rad-antibodies.com/protocols. Yes No Not Determined Suggested Dilution Western Blotting 1/1000 PrecisionAb antibodies have been extensively validated for the western blot application. The antibody has been validated at the suggested dilution. Where this product has not been tested for use in a particular technique this does not necessarily exclude its use in such procedures. Further optimization may be required dependant on sample type. Target Species Human Species Cross Reacts with: Mouse, Rat Reactivity N.B. Antibody reactivity and working conditions may vary between species. Product Form Purified IgG - liquid Preparation 20μl Rabbit polyclonal antibody purified by affinity chromatography Buffer Solution Phosphate buffered saline Preservative 0.09% Sodium Azide (NaN ) Stabilisers 3 Immunogen KLH-conjugated synthetic peptide corresponding to aa 4-33 of human PRPF19 External Database Links UniProt: Q9UMS4 Related reagents Entrez Gene: 27339 PRPF19 Related reagents Synonyms NMP200, PRP19, SNEV Page 1 of 3 Specificity Rabbit anti Human PRPF19 antibody recognizes PRPF19, also known as PRP19/PSO4 homolog, PRP19/PSO4 pre-mRNA processing factor 19 homolog, nuclear matrix protein 200, nuclear matrix protein NMP200 related to splicing factor PRP19, psoralen 4 and senescence evasion factor. The PRPF19 gene is the human homolog of yeast Pso4, a gene essential for cell survival and DNA repair (Beck et al. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Runx1 Gene in Acute Myeloid Leukemia: Expression Level Signifcance and Impact on Clinical Features
Runx1 Gene in Acute Myeloid Leukemia: Expression Level Signicance and Impact on Clinical Features Fadwa Said Abdelazim Mohamed ( [email protected] ) Cairo University Roxan E Shak National Cancer Institute, Cairo University Naglaa M. Hassan National Cancer Institute, Cairo University Research article Keywords: Acute myeloid leukemia, RUNX1, mutation, gene expression, Egypt Posted Date: April 7th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-20926/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/20 Abstract Background Acute myeloid leukemia represents the highest percentage of all adult acute leukemia variants. Runt-related transcription factor1 (RUNX1), a transcription factor with a known tumor suppressor function was recently reported as a tumor promotor in AML. We investigated the role of RUNX1 geneexpression levelin Egyptian AML patients and delineateits clinical signicance. Methods This study recruited 91 AML patients that were recently diagnosed at our hospital with 14 healthy age- and sex-matched donors of bone marrow transplantation unit. We measured RUNX1 gene expression level using reverse transcription–quantitative polymerase chain reaction. Results We found that RUNX1 gene expression level was signicantly higher compared to the control group (p < 0.001). Patients with FLT3 mutations had the higher expression level of RUNX1 (p=0.023). The male patients expressed signicantly higher level of RUNX1 (p=0.046). Conclusion The RUNX1 gene expression may serve as a diagnostic marker in Egyptian AML patients. Its relation to FLT3 may give clue that patients carrying this mutation may benet fromnew treatments that target RUNX1 in the future. -

Nuclear PTEN Safeguards Pre-Mrna Splicing to Link Golgi Apparatus for Its Tumor Suppressive Role
ARTICLE DOI: 10.1038/s41467-018-04760-1 OPEN Nuclear PTEN safeguards pre-mRNA splicing to link Golgi apparatus for its tumor suppressive role Shao-Ming Shen1, Yan Ji2, Cheng Zhang1, Shuang-Shu Dong2, Shuo Yang1, Zhong Xiong1, Meng-Kai Ge1, Yun Yu1, Li Xia1, Meng Guo1, Jin-Ke Cheng3, Jun-Ling Liu1,3, Jian-Xiu Yu1,3 & Guo-Qiang Chen1 Dysregulation of pre-mRNA alternative splicing (AS) is closely associated with cancers. However, the relationships between the AS and classic oncogenes/tumor suppressors are 1234567890():,; largely unknown. Here we show that the deletion of tumor suppressor PTEN alters pre-mRNA splicing in a phosphatase-independent manner, and identify 262 PTEN-regulated AS events in 293T cells by RNA sequencing, which are associated with significant worse outcome of cancer patients. Based on these findings, we report that nuclear PTEN interacts with the splicing machinery, spliceosome, to regulate its assembly and pre-mRNA splicing. We also identify a new exon 2b in GOLGA2 transcript and the exon exclusion contributes to PTEN knockdown-induced tumorigenesis by promoting dramatic Golgi extension and secretion, and PTEN depletion significantly sensitizes cancer cells to secretion inhibitors brefeldin A and golgicide A. Our results suggest that Golgi secretion inhibitors alone or in combination with PI3K/Akt kinase inhibitors may be therapeutically useful for PTEN-deficient cancers. 1 Department of Pathophysiology, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, Shanghai Jiao Tong University School of Medicine (SJTU-SM), Shanghai 200025, China. 2 Institute of Health Sciences, Shanghai Institutes for Biological Sciences of Chinese Academy of Sciences and SJTU-SM, Shanghai 200025, China. -
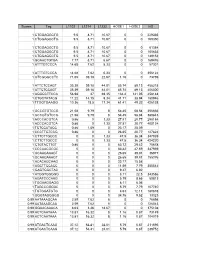
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 229335 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 169350 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 61384 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 105633 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 149152 1 GCAACTGTGA 7.77 8.71 6.67 0 0 169476 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 57301 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 356122 1 GTCGGGCCTC 71.65 39.18 22.67 1.16 0 73769 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 458218 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 406300 1 AGGGCTTCCA 56.98 37 69.35 134.4 141.05 458148 1 CTGCTATACG 11.22 14.15 9.34 41.71 38.94 180946 1 TTGGTGAAGG 10.36 18.5 17.34 61.41 49.32 426138 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 356666 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 380843 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 298184 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 400124 1 TCTCCATACC 0.86 1.09 0 23.17 25.09 1 CCCTTGTCCG 0.86 0 0 26.65 20.77 127824 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 347939 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 424220 1 CTGTACTTGT 0.86 0 0 63.72 29.42 75678 1 CCCAACGCGC 0 0 0 83.42 47.59 347939 1 GCAAGAAAGT 0 0 0 26.65 39.81 36977 1 GCAAGAAAGT 0 0 0 26.65 39.81 155376 1 ACACAGCAAG 0 0 0 23.17 15.58 1 AGCTTCCACC 0 0 0 11.59 7.79 355542 1 GAGTGGCTAC 0 0 0 9.27 6.92 1 ATGGTGGGGG 0 0 0 8.11 22.5 343586 1 AGATCCCAAG 0 0 0 5.79 8.65 50813 1 TGGAAGGAGG 0 0 0 8.11 6.06 1 TAGCCGGGAC 0 0 0 5.79 7.79 107740 1 TGTGGATGTG 0 0 0 4.63 12.11 180878 1 GGGTAGGGGG 0 0 0 34.76 9.52 13323 0.99 AATAAAGCAA 2.59 7.62 8 0 0 76698 0.99 AATAAAGCAA 2.59 7.62 8 0 0 126043 0.99 GGAACAAACA 8.63 3.26 18.67 0 0 375108 -

(12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer Et Al
USOO8440393B2 (12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer et al. (45) Date of Patent: May 14, 2013 (54) PRO-ANGIOGENIC GENES IN OVARIAN OTHER PUBLICATIONS TUMORENDOTHELIAL CELL, SOLATES Boyd (The Basic Science of Oncology, 1992, McGraw-Hill, Inc., p. (75) Inventors: Michael J. Birrer, Mt. Airy, MD (US); 379). Tomas A. Bonome, Washington, DC Tockman et al. (Cancer Res., 1992, 52:2711s-2718s).* (US); Anil Sood, Pearland, TX (US); Pritzker (Clinical Chemistry, 2002, 48: 1147-1150).* Chunhua Lu, Missouri City, TX (US) Benedict et al. (J. Exp. Medicine, 2001, 193(1) 89-99).* Jiang et al. (J. Biol. Chem., 2003, 278(7) 4763-4769).* (73) Assignees: The United States of America as Matsushita et al. (FEBS Letters, 1999, vol. 443, pp. 348-352).* Represented by the Secretary of the Singh et al. (Glycobiology, 2001, vol. 11, pp. 587-592).* Department of Health and Human Abbosh et al. (Cancer Res. Jun. 1, 2006 66:5582-55.91 and Supple Services, Washington, DC (US); The mental Figs. S1-S7).* University of MD Anderson Cancer Zhai et al. (Chinese General Practice Aug. 2008, 11(8A): 1366 Center, Houston, TX (US) 1367).* Lu et al. (Cancer Res. Feb. 15, 2007, 64(4): 1757-1768).* (*) Notice: Subject to any disclaimer, the term of this Bagnato et al., “Activation of Mitogenic Signaling by Endothelin 1 in patent is extended or adjusted under 35 Ovarian Carcinoma Cells', Cancer Research, vol. 57, pp. 1306-1311, U.S.C. 154(b) by 194 days. 1997. Bouras et al., “Stanniocalcin 2 is an Estrogen-responsive Gene (21) Appl. -

Human Prefoldin Modulates Co-Transcriptional Pre-Mrna Splicing
bioRxiv preprint doi: https://doi.org/10.1101/2020.06.14.150466; this version posted July 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. BIOLOGICAL SCIENCES: Biochemistry Human prefoldin modulates co-transcriptional pre-mRNA splicing Payán-Bravo L 1,2, Peñate X 1,2 *, Cases I 3, Pareja-Sánchez Y 1, Fontalva S 1,2, Odriozola Y 1,2, Lara E 1, Jimeno-González S 2,5, Suñé C 4, Reyes JC 5, Chávez S 1,2. 1 Instituto de Biomedicina de Sevilla, Universidad de Sevilla-CSIC-Hospital Universitario V. del Rocío, Seville, Spain. 2 Departamento de Genética, Facultad de Biología, Universidad de Sevilla, Seville, Spain. 3 Centro Andaluz de Biología del Desarrollo, CSIC-Universidad Pablo de Olavide, Seville, Spain. 4 Department of Molecular Biology, Institute of Parasitology and Biomedicine "López Neyra" IPBLN-CSIC, PTS, Granada, Spain. 5 Andalusian Center of Molecular Biology and Regenerative Medicine-CABIMER, Junta de Andalucia-University of Pablo de Olavide-University of Seville-CSIC, Seville, Spain. Correspondence: Sebastián Chávez, IBiS, campus HUVR, Avda. Manuel Siurot s/n, Sevilla, 41013, Spain. Phone: +34-955923127: e-mail: [email protected]. * Co- corresponding; [email protected]. bioRxiv preprint doi: https://doi.org/10.1101/2020.06.14.150466; this version posted July 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract Prefoldin is a heterohexameric complex conserved from archaea to humans that plays a cochaperone role during the cotranslational folding of actin and tubulin monomers. -

Mrna Editing, Processing and Quality Control in Caenorhabditis Elegans
| WORMBOOK mRNA Editing, Processing and Quality Control in Caenorhabditis elegans Joshua A. Arribere,*,1 Hidehito Kuroyanagi,†,1 and Heather A. Hundley‡,1 *Department of MCD Biology, UC Santa Cruz, California 95064, †Laboratory of Gene Expression, Medical Research Institute, Tokyo Medical and Dental University, Tokyo 113-8510, Japan, and ‡Medical Sciences Program, Indiana University School of Medicine-Bloomington, Indiana 47405 ABSTRACT While DNA serves as the blueprint of life, the distinct functions of each cell are determined by the dynamic expression of genes from the static genome. The amount and specific sequences of RNAs expressed in a given cell involves a number of regulated processes including RNA synthesis (transcription), processing, splicing, modification, polyadenylation, stability, translation, and degradation. As errors during mRNA production can create gene products that are deleterious to the organism, quality control mechanisms exist to survey and remove errors in mRNA expression and processing. Here, we will provide an overview of mRNA processing and quality control mechanisms that occur in Caenorhabditis elegans, with a focus on those that occur on protein-coding genes after transcription initiation. In addition, we will describe the genetic and technical approaches that have allowed studies in C. elegans to reveal important mechanistic insight into these processes. KEYWORDS Caenorhabditis elegans; splicing; RNA editing; RNA modification; polyadenylation; quality control; WormBook TABLE OF CONTENTS Abstract 531 RNA Editing and Modification 533 Adenosine-to-inosine RNA editing 533 The C. elegans A-to-I editing machinery 534 RNA editing in space and time 535 ADARs regulate the levels and fates of endogenous dsRNA 537 Are other modifications present in C. -

Functional Analysis of the Nuclear Basket and Protein Tpr
Haruki Iino (Autor) Functional analysis of the nuclear basket and protein Tpr https://cuvillier.de/de/shop/publications/7587 Copyright: Cuvillier Verlag, Inhaberin Annette Jentzsch-Cuvillier, Nonnenstieg 8, 37075 Göttingen, Germany Telefon: +49 (0)551 54724-0, E-Mail: [email protected], Website: https://cuvillier.de 2. Abstract A large coiled-coil protein, Tpr (Translocated promoter region) is located at the nuclear side of the nuclear pore complex (NPC), and plays an important role in the architecture of the nuclear basket (NB). Although its contribution to the NB structure has been characterized, little is known about the function of Tpr. In this work, we investigated whether and how Tpr contributes to the surveillance of mRNA export, especially, to the quality control (QC) of certain un-spliced mRNAs. To answer these questions, we established a series of different reporter cell lines, allowing us to monitor the occurrence of un-spliced and spliced reporter transcripts. In the analysis of most of these reporter cell lines, the depletion of Tpr did not cause any obvious leakage of un-spliced reporter mRNAs. However, when the coding sequence of the HIV (Human immunodeficiency virus) Gag protein was used as a readout, the cytoplasmic levels of the un-spliced HIV-gag mRNAs were highly enhanced in the absence of Tpr. Such phenotype was only observed when a viral RNA export enhancer element, the CTE (the constitutive transport element), that recruits the general mRNA export receptor, NXF1/TAP, was part of the HIV-gag reporter gene. Intriguingly, a slight reduction in the cellular Tpr levels already caused the breakdown of a retention mechanism that normally keeps these HIV-gag transcripts in the nucleus. -

Candidate Genes, Pathways and Mechanisms for Bipolar (Manic–Depressive) and Related Disorders: an Expanded Convergent Function
Molecular Psychiatry (2004), 1007–1029 & 2004 Nature Publishing Group All rights reserved 1359-4184/04 $30.00 www.nature.com/mp ORIGINAL RESEARCH ARTICLE Candidate genes, pathways and mechanisms for bipolar (manic–depressive) and related disorders: an expanded convergent functional genomics approach CA Ogden1,2,3, ME Rich1,2,3, NJ Schork2, MP Paulus2,3, MA Geyer2,3, JB Lohr2,3, R Kuczenski2,3 and AB Niculescu1,2,3 1Laboratory of Neurophenomics, University of California, San Diego, CA, USA; 2Department of Psychiatry, University of California, San Diego, CA, USA; 3VA San Diego Healthcare System VISN-22 MIRECC, San Diego, CA, USA Identifying genes for bipolar mood disorders through classic genetics has proven difficult. Here, we present a comprehensive convergent approach that translationally integrates brain gene expression data from a relevant pharmacogenomic mouse model (involving treatments with a stimulant—methamphetamine, and a mood stabilizer—valproate), with human data (linkage loci from human genetic studies, changes in postmortem brains from patients), as a bayesian strategy of crossvalidating findings. Topping the list of candidate genes, we have DARPP-32 (dopamine- and cAMP-regulated phosphoprotein of 32 kDa) located at 17q12, PENK (preproenkephalin) located at 8q12.1, and TAC1 (tachykinin 1, substance P) located at 7q21.3. These data suggest that more primitive molecular mechanisms involved in pleasure and pain may have been recruited by evolution to play a role in higher mental functions such as mood. The analysis also revealed other high-probability candidates genes (neurogenesis, neuro- trophic, neurotransmitter, signal transduction, circadian, synaptic, and myelin related), pathways and mechanisms of likely importance in pathophysiology. -

(12) Patent Application Publication (10) Pub. No.: US 2013/0274315 A1 Birrer Et Al
US 201302743 15A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2013/0274315 A1 Birrer et al. (43) Pub. Date: Oct. 17, 2013 (54) PRO-ANGIOGENC GENES IN OVARAN (60) Provisional application No. 60/901,455, filed on Feb. TUMORENDOTHELIAL CELL, SOLATES 14, 2007. (71) Applicants: The University of Texas MD Anderson Publication Classification Cancer Center, Houston, TX (US); The Government of the U.S.A as (51) Int. Cl. represented by the Secretary of the CI2O I/68 (2006.01) Department of He, Rockville, MD (US) (52) U.S. Cl. CPC .................................... CI2O I/6886 (2013.01) (72) Inventors: Michael J. Birrer, Mt. Airy, MD (US); USPC ............... 514/44A: 435/6.12: 506/9: 435/7.1 Tomas A. Bonome, Washington, DC (US); Anil Sood, Pearland, TX (US); (57) ABSTRACT Chunhua Lu, Missouri City, TX (US) A gene profiling signature for ovarian tumor endothelial cells is disclosed herein. The gene signature can be used to diag nosis or prognosis an ovarian tumor, identify agents to treat an (21) Appl. No.: 13/863,219 ovariantumor, to predict the metastatic potential of an ovarian tumor and to determine the effectiveness of ovarian tumor (22) Filed: Apr. 15, 2013 treatments. Thus, methods are provided for identifying agents that can be used to treat ovarian cancer, for determining the effectiveness of an ovarian tumor treatment, or to diagnose or Related U.S. Application Data prognose an ovarian tumor. Methods of treatment are also (60) Division of application No. 12/541,729, filed on Aug. disclosed which include administering a composition that 14, 2009, now Pat.