Vaccinia Virus As a Master of Host Shutoff Induction: Targeting Processes of the Central Dogma and Beyond
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Enhanced Expression of Vascular Endothelial Growth Factor (VEGF) Plays a Critical Role in the Tumor Progression Potential Induced by Simian Virus 40 Large T Antigen
Oncogene (2002) 21, 2896 ± 2900 ã 2002 Nature Publishing Group All rights reserved 0950 ± 9232/02 $25.00 www.nature.com/onc Enhanced expression of vascular endothelial growth factor (VEGF) plays a critical role in the tumor progression potential induced by simian virus 40 large T antigen Alfonso Catalano1, Mario Romano*,2, Stefano Martinotti3 and Antonio Procopio*,1 1Institute of Experimental Pathology, University of Ancona, Faculty of Medicine, Via Ranieri, 60131 Ancona, Italy; 2Department of Human Pathology, University of Messina, 98125 Messina, Italy; 3Department of Oncology and Neuroscience, `G. D'Annunzio' University, Chieti, Italy Vascular endothelial growth factor (VEGF), an impor- disorders, including mesotheliomas (Kumar-Singh et tant angiogenic factor, regulates cell proliferation, al., 1999). Among these, VEGF, whose crucial role in dierentiation, and apoptosis through activation of its tumor angiogenesis is well established (Hanahan and tyrosine-kinase receptors, such as Flt-1 and Flk-1/Kdr. Folkman., 1996), acts also as a potent mitogen and Human malignant mesothelioma cells (HMC), which survival factor for human malignant mesothelioma have wild-type p53, express VEGF and exhibit cell cells (HMC). In fact, VEGF regulates HMC prolifera- growth increased by VEGF. Here, we demonstrate that tion through its tyrosine-kinase receptors activation early transforming proteins of simian virus (SV) 40, large (i.e. Flt-1 and KDR/¯k-1) (Strizzi et al., 2001). In tumor antigen (Tag) and small tumor antigen (tag), addition, VEGF is the main eector of 5-lipoxygenase which have been associated with mesotheliomas, en- action on HMC survival (Romano et al., 2001). Thus, hanced HMC proliferation by inducing VEGF expres- VEGF plays a key role by regulating multiple sion. -

A Polyoma Mutant That Encodessmall T Antigen but Not Middle T Antigen
MOLECULAR AND CELLULAR BIOLOGY, Dec. 1984, p. 2774-2783 Vol. 4, No. 12 0270-7306/84/122774-10$02.00/0 Copyright © 1984, American Society for Microbiology A Polyoma Mutant That Encodes Small T Antigen But Not Middle T Antigen Demonstrates Uncoupling of Cell Surface and Cytoskeletal Changes Associated with Cell Transformation T. JAKE LIANG, GORDON G. CARMICHAEL,t AND THOMAS L. BENJAMIN* Department of Pathology, Harvard Medical School, Boston, Massachusetts 02115 Received 2 July 1984/Accepted 31 August 1984 The hr-t gene of polyoma virus encodes both the small and middle T (tumor) antigens and exerts pleiotropic effects on cells. By mutating the 3' splice site for middle T mRNA, we have constructed a virus mutant, Py8O8A, which fails to express middle T but encodes normal small and large T proteins. The mutant failed to induce morphological transformation or growth in soft agar, but did stimulate postconfluent growth of normal cells. Cells infected by Py8O8A became fully agglutinable by lectins while retaining normal actin cable architecture and normal levels of extracellular fibronectin. These properties of Py8O8A demonstrated the separability of structural changes at the cell surface from those in the cytoskeleton and extracellular matrix, parameters which have heretofore been linked in the action of the hr-t and other viral oncogenes. The early region of polyoma encodes three proteins de- substitution of phenylalanine for tyrosine at position 315 in tected as tumor (T) antigens. These polypeptides of 100,000 middle T has been made and studied in two laboratories, daltons (100K; large T), 56K (middle T), and 22K (small T) with different results. -

Runx1 Gene in Acute Myeloid Leukemia: Expression Level Signifcance and Impact on Clinical Features
Runx1 Gene in Acute Myeloid Leukemia: Expression Level Signicance and Impact on Clinical Features Fadwa Said Abdelazim Mohamed ( [email protected] ) Cairo University Roxan E Shak National Cancer Institute, Cairo University Naglaa M. Hassan National Cancer Institute, Cairo University Research article Keywords: Acute myeloid leukemia, RUNX1, mutation, gene expression, Egypt Posted Date: April 7th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-20926/v1 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Page 1/20 Abstract Background Acute myeloid leukemia represents the highest percentage of all adult acute leukemia variants. Runt-related transcription factor1 (RUNX1), a transcription factor with a known tumor suppressor function was recently reported as a tumor promotor in AML. We investigated the role of RUNX1 geneexpression levelin Egyptian AML patients and delineateits clinical signicance. Methods This study recruited 91 AML patients that were recently diagnosed at our hospital with 14 healthy age- and sex-matched donors of bone marrow transplantation unit. We measured RUNX1 gene expression level using reverse transcription–quantitative polymerase chain reaction. Results We found that RUNX1 gene expression level was signicantly higher compared to the control group (p < 0.001). Patients with FLT3 mutations had the higher expression level of RUNX1 (p=0.023). The male patients expressed signicantly higher level of RUNX1 (p=0.046). Conclusion The RUNX1 gene expression may serve as a diagnostic marker in Egyptian AML patients. Its relation to FLT3 may give clue that patients carrying this mutation may benet fromnew treatments that target RUNX1 in the future. -

Review Article Human Polyomaviruses in Skin Diseases
SAGE-Hindawi Access to Research Pathology Research International Volume 2011, Article ID 123491, 12 pages doi:10.4061/2011/123491 Review Article Human Polyomaviruses in Skin Diseases Ugo Moens,1 Maria Ludvigsen,1 and Marijke Van Ghelue2 1 Institute of Medical Biology, Faculty of Health Sciences, University of Tromsø, 9037 Tromsø, Norway 2 Department of Medical Genetics, University Hospital of Northern-Norway, 9038 Tromsø, Norway Correspondence should be addressed to Ugo Moens, [email protected] Received 28 February 2011; Accepted 29 June 2011 Academic Editor: Gerardo Ferrara Copyright © 2011 Ugo Moens et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Polyomaviruses are a family of small, nonenveloped viruses with a circular double-stranded DNA genome of ∼5,000 base pairs protected by an icosahedral protein structure. So far, members of this family have been identified in birds and mammals. Until 2006, BK virus (BKV), JC virus (JCV), and simian virus 40 (SV40) were the only polyomaviruses known to circulate in the human population. Their occurrence in individuals was mainly confirmed by PCR and the presence of virus-specific antibodies. Using the same methods, lymphotropic polyomavirus, originally isolated in monkeys, was recently shown to be present in healthy individuals although with much lower incidence than BKV, JCV, and SV40. The use of advanced high-throughput sequencing and improved rolling circle amplification techniques have identified the novel human polyomaviruses KI, WU, Merkel cell polyomavirus, HPyV6, HPyV7, trichodysplasia spinulosa-associated polyomavirus, and HPyV9. -

(12) Patent Application Publication (10) Pub. No.: US 2004/0023207 A1 Polansky (43) Pub
US 20040023207A1 (19) United States (12) Patent Application Publication (10) Pub. No.: US 2004/0023207 A1 Polansky (43) Pub. Date: Feb. 5, 2004 (54) ASSAYS FOR DRUG DISCOVERY BASED ON Publication Classification MICROCOMPETITION WITH A FOREIGN POLYNUCLEOTIDE (51) Int. Cl." .............................. C12O 1/70; C12O 1/68; C12N 15/85; C12N 15/86 (52) U.S. Cl. .................. 435/5; 435/6; 435/455; 435/456 (76) Inventor: Hanan Polansky, Rochester, NY (US) (57) ABSTRACT Correspondence Address: A recent discovery showed that microcompetition between a Hanan Polansky foreign polynucleotide and a cellular polynucleotide is a risk 3159 S. Winton Rd. factor for Some of the major chronic diseases. The invention Rochester, NY 14623 (US) uses this novel discovery to present assays for Screening compounds based on their effectiveness in modulating Such microcompetition. The effective compounds can be used in (21) Appl. No.: 10/211,295 treatment of these chronic diseases. The invention also presents assays for Screening compounds that can be used in treatment of chronic diseaseS resulting from other foreign (22) Filed: Aug. 1, 2002 polynucleotide-type disruptions. Patent Application Publication Feb. 5, 2004 Sheet 1 of 6 US 2004/0023207 A1 s e 2 9 s H c 2 4 competitor plasmid/test plasmid Figure 1 1 O 09% 1 2 3 4 competitor plasmid/test plasmid -H Ltk- (pSV2Neo) -A-ML (pSV2neo) . A- - - Ltk- (pA1 Oneo) - - - - - - Mill (pA1 Oneo) Figure 2 Patent Application Publication Feb. 5, 2004 Sheet 2 of 6 US 2004/0023207 A1 e 2 r U H c O s S 2 3. Molar ratio (pSV2NeolhMT-ilA-CAT) Figure 3 Patent Application Publication Feb. -

Tumor Antigen (Hybrid Trp-Lac Promoter/Actin Cable Effect) ILAN BIKEL*, THOMAS M
Proc. NatL Acad. Sci. USA Vol. 80, pp. 906-910, February 1983 Biochemistry Purification of biologically active simian virus 40 small tumor antigen (hybrid trp-lac promoter/actin cable effect) ILAN BIKEL*, THOMAS M. ROBERTS*, MARILUCI T. BLADON*, ROBERT GREEN*, EGON AMANNt, AND DAVID M. LIVINGSTON* *The Sidney Farber Cancer Institute and The Departments of Medicine and Pathology, Harvard Medical School, Boston, Massachusetts 02115; and tDepartment of Biochemistry and Molecular Biology, Harvard University, Cambridge, Massachusetts 02138 Communicated by Morris Friedkin, October 29, 1982 The simian virus 40 small tumor antigen (t antigen) gene has been were grown in L broth containing ampicillin (40 Ag/ml) to an cloned downstream from a hybrid Escherichia coli trp-lac pro- ODW of0.7 and then incubated for 2 hr with 5 mM isopropyl moter anda suitable ribosome binding site. Abacterial clone (865i) f3-D-thiogalactopyranoside. Radiolabeling of proteins with transformed by such a plasmid (pTR865) expresses this gene and, [3S]methionine was performed as described (31). under optimal conditions, can produce .5% ofits total protein as t Antigen and Protein Assays. t antigen was detected by Na- t antigen. Soluble extracts ofsuch a clone were relatively depleted DodSO4/polyacrylamide slab gel electrophoresis (32) using 4% in t antigen, which was found in the initial pellet fraction. The stacking and 15% separating gels. Marker proteins were bovine protein was recovered from this fraction in a significantly purified serum albumin (Mr 68,000), carbonic anhydrase (29,000), form byextraction with urea-containing buffer. After gel filtration concentrations were deter- of such t antigen-enriched solutions, highly purified protein was and myoglobin (17,000). -

PP2A-Dependent Disruption of Centrosome Replication and Cytoskeleton Organization in Drosophila by SV40 Small Tumor Antigen
Oncogene (2008) 27, 6334–6346 & 2008 Macmillan Publishers Limited All rights reserved 0950-9232/08 $32.00 www.nature.com/onc ORIGINAL ARTICLE PP2A-dependent disruption of centrosome replication and cytoskeleton organization in Drosophila by SV40 small tumor antigen S Kotadia1,LRKao1, SA Comerford2, RT Jones1, RE Hammer3 and TL Megraw1 1Department of Pharmacology, The Cecil and Ida Green Center for Reproductive Biology Sciences, The University of Texas Southwestern Medical Center at Dallas, Dallas, TX, USA; 2Department of Molecular Genetics, The Cecil and Ida Green Center for Reproductive Biology Sciences, The University of Texas Southwestern Medical Center at Dallas, Dallas, TX, USA and 3Department of Biochemistry, The Cecil and Ida Green Center for Reproductive Biology Sciences, The University of Texas Southwestern Medical Center at Dallas, Dallas, TX, USA Viruses of the DNA tumor virus family share the ability to cell-cycle and apoptosis regulation, viral oncoproteins transform vertebrate cells through the action of virus- transform cells. Investigation of DNA tumor virus encoded tumor antigens that interfere with normal cell oncoproteins has led to the identification of many physiology. They accomplish this very efficiently by fundamental mechanisms of tumor suppression (Lavia inhibiting endogenous tumor suppressor proteins that et al., 2003; Ahuja et al., 2005). By altering the activity control cell proliferation and apoptosis. Simian virus 40 of p53, retinoblastoma protein and serine/threonine (SV40) encodes two oncoproteins, large tumor antigen, protein phosphatase 2A (PP2A), three key tumor which directly inhibits the tumor suppressors p53 and Rb, suppressors, SV40 can cause tumor formation in and small tumor antigen (ST), which interferes with transgenic mouse models (Ahuja et al., 2005; Arroyo serine/threonine protein phosphatase 2A (PP2A). -
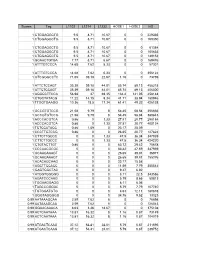
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1
Scores Tag L1102 L1214 L1232 HOSE1 HOSE2 HS 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 229335 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 169350 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 61384 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 105633 1 CTGGAGGCTG 9.5 8.71 10.67 0 0 149152 1 GCAACTGTGA 7.77 8.71 6.67 0 0 169476 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 57301 1 ATTTGTCCCA 14.68 7.62 5.33 0 0 356122 1 GTCGGGCCTC 71.65 39.18 22.67 1.16 0 73769 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 458218 1 ATTCTCCAGT 35.39 39.18 44.01 85.74 89.13 406300 1 AGGGCTTCCA 56.98 37 69.35 134.4 141.05 458148 1 CTGCTATACG 11.22 14.15 9.34 41.71 38.94 180946 1 TTGGTGAAGG 10.36 18.5 17.34 61.41 49.32 426138 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 356666 1 GCCGTGTCCG 21.58 9.79 8 54.45 58.84 380843 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 298184 1 ACCCACGTCA 0.86 0 1.33 27.81 20.77 400124 1 TCTCCATACC 0.86 1.09 0 23.17 25.09 1 CCCTTGTCCG 0.86 0 0 26.65 20.77 127824 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 347939 1 CTTCTTGCCC 0 0 1.33 47.5 36.34 424220 1 CTGTACTTGT 0.86 0 0 63.72 29.42 75678 1 CCCAACGCGC 0 0 0 83.42 47.59 347939 1 GCAAGAAAGT 0 0 0 26.65 39.81 36977 1 GCAAGAAAGT 0 0 0 26.65 39.81 155376 1 ACACAGCAAG 0 0 0 23.17 15.58 1 AGCTTCCACC 0 0 0 11.59 7.79 355542 1 GAGTGGCTAC 0 0 0 9.27 6.92 1 ATGGTGGGGG 0 0 0 8.11 22.5 343586 1 AGATCCCAAG 0 0 0 5.79 8.65 50813 1 TGGAAGGAGG 0 0 0 8.11 6.06 1 TAGCCGGGAC 0 0 0 5.79 7.79 107740 1 TGTGGATGTG 0 0 0 4.63 12.11 180878 1 GGGTAGGGGG 0 0 0 34.76 9.52 13323 0.99 AATAAAGCAA 2.59 7.62 8 0 0 76698 0.99 AATAAAGCAA 2.59 7.62 8 0 0 126043 0.99 GGAACAAACA 8.63 3.26 18.67 0 0 375108 -

(12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer Et Al
USOO8440393B2 (12) United States Patent (10) Patent No.: US 8.440,393 B2 Birrer et al. (45) Date of Patent: May 14, 2013 (54) PRO-ANGIOGENIC GENES IN OVARIAN OTHER PUBLICATIONS TUMORENDOTHELIAL CELL, SOLATES Boyd (The Basic Science of Oncology, 1992, McGraw-Hill, Inc., p. (75) Inventors: Michael J. Birrer, Mt. Airy, MD (US); 379). Tomas A. Bonome, Washington, DC Tockman et al. (Cancer Res., 1992, 52:2711s-2718s).* (US); Anil Sood, Pearland, TX (US); Pritzker (Clinical Chemistry, 2002, 48: 1147-1150).* Chunhua Lu, Missouri City, TX (US) Benedict et al. (J. Exp. Medicine, 2001, 193(1) 89-99).* Jiang et al. (J. Biol. Chem., 2003, 278(7) 4763-4769).* (73) Assignees: The United States of America as Matsushita et al. (FEBS Letters, 1999, vol. 443, pp. 348-352).* Represented by the Secretary of the Singh et al. (Glycobiology, 2001, vol. 11, pp. 587-592).* Department of Health and Human Abbosh et al. (Cancer Res. Jun. 1, 2006 66:5582-55.91 and Supple Services, Washington, DC (US); The mental Figs. S1-S7).* University of MD Anderson Cancer Zhai et al. (Chinese General Practice Aug. 2008, 11(8A): 1366 Center, Houston, TX (US) 1367).* Lu et al. (Cancer Res. Feb. 15, 2007, 64(4): 1757-1768).* (*) Notice: Subject to any disclaimer, the term of this Bagnato et al., “Activation of Mitogenic Signaling by Endothelin 1 in patent is extended or adjusted under 35 Ovarian Carcinoma Cells', Cancer Research, vol. 57, pp. 1306-1311, U.S.C. 154(b) by 194 days. 1997. Bouras et al., “Stanniocalcin 2 is an Estrogen-responsive Gene (21) Appl. -

Interactions Between Polyomavirus Large T Antigen and the Viral Replication Origin Dna: How and Why
INTERACTIONS BETWEEN POLYOMAVIRUS LARGE T ANTIGEN AND THE VIRAL REPLICATION ORIGIN DNA: HOW AND WHY by Yu-Cai Peng Department of Microbiology and Immunology McGill University, Montreal April, 1999 A thesis submitted to the Faculty of Craduate Studies and Re3earch in partial fulfullmeat of the requirements of the degree of doctor of philosophy @Yu-Cai Peng, 1999 National Library Biblioth ue nationale 1+1 of Canada du Cana7 a Acquisitions and Acquisitions et Bibliographie Srvices services bibliographiques 395 Wellmgton Street 395, nie Weliingîm Ottawa ON KIA ON4 OrtawaON K1AON4 Canada Canada The author has granted a non- L'auteur a accordé une licence non exclusive Licence ailowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, distribute or sell reproduire, prêter, distribuer ou copies of this thesis in microform, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfichelfilm, de reproduction sur papier ou sur format électronique. The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fkom it Ni la thèse ni des extraits substantiels may be printed or othenvise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation. TABLE OF CONTENTS Page Tableofcontents................................................... I Abstract .......................................................... VI Resumé........................................................... VI11 Acknowledgements ................................................. X Claim of contribution to kaowledge ................................... XI Listoffigures ...................................................... XllI Guidelines regarding doctoral thesis ................................... XV CHAPTER 1. INTRODUCTION .................................... 1 1. Overview: Life cycle of polyomavirus and simian virus 40 .......... -

Polyoma Virus Small Tumor Antigen Pre-Mrna Splicing Requires Cooperation Between Two 3' Splice Sites Hui GE, JONATHAN NOBLE, JOHN COLGAN, and JAMES L
Proc. Nati. Acad. Sci. USA Vol. 87, pp. 3338-3342, May 1990 Biochemistry Polyoma virus small tumor antigen pre-mRNA splicing requires cooperation between two 3' splice sites Hui GE, JONATHAN NOBLE, JOHN COLGAN, AND JAMES L. MANLEY Department of Biological Sciences, Columbia University, New York, NY 10027 Communicated by Joan A. Steitz, January 25, 1990 (receivedfor review December 13, 1989) ABSTRACT We have studied splicing ofthe polyoma virus proteins also likely play a role in the splicing reaction (e.g., early region pre-mRNA in vitro. This RNA is alternatively refs. 19-22). spliced in vivo to produce mRNA encoding the large, middle- The complexity and size of the splicing apparatus suggests sized (MTAg), and small (StAg) tumor antigens. Our primary that steric constraints may play a role in splicing of some interest was to learn how the 48-nucleotide StAg intron is pre-mRNAs. Consistent with this proposal, several studies excised, because the length of this intron is significantly less have shown that introns in higher eukaryotes have a mini- than the apparent minimum established for mammalian in- mum size requirement. Roughly 45 nucleotides (nt) must trons. Although the products ofall three splices are detected in separate the 5' splice site and branch point (23-27), and the vitro, characterization of the pathway and sequence require- minimum distance between the branch point and 3' splice site ments of StAg splicing suggests that splicing factors interact appears to be approximately 18 nt (26, 28). The simian virus with the precursor RNA in an unexpected way to catalyze 40 small tumor antigen pre-mRNA contains an intron of66 nt removal ofthis intron. -

Drosophila As a Model for Infectious Diseases
International Journal of Molecular Sciences Review Drosophila as a Model for Infectious Diseases J. Michael Harnish 1,2 , Nichole Link 1,2,3,† and Shinya Yamamoto 1,2,4,5,* 1 Department of Molecular and Human Genetics, Baylor College of Medicine (BCM), Houston, TX 77030, USA; [email protected] (J.M.H.); [email protected] (N.L.) 2 Jan and Dan Duncan Neurological Research Institute, Texas Children’s Hospital, Houston, TX 77030, USA 3 Howard Hughes Medical Institute, Houston, TX 77030, USA 4 Department of Neuroscience, BCM, Houston, TX 77030, USA 5 Development, Disease Models and Therapeutics Graduate Program, BCM, Houston, TX 77030, USA * Correspondence: [email protected]; Tel.: +1-832-824-8119 † Current Affiliation: Department of Neurobiology, University of Utah, Salt Lake City, UT 84112, USA. Abstract: The fruit fly, Drosophila melanogaster, has been used to understand fundamental principles of genetics and biology for over a century. Drosophila is now also considered an essential tool to study mechanisms underlying numerous human genetic diseases. In this review, we will discuss how flies can be used to deepen our knowledge of infectious disease mechanisms in vivo. Flies make effective and applicable models for studying host-pathogen interactions thanks to their highly con- served innate immune systems and cellular processes commonly hijacked by pathogens. Drosophila researchers also possess the most powerful, rapid, and versatile tools for genetic manipulation in multicellular organisms. This allows for robust experiments in which specific pathogenic proteins can be expressed either one at a time or in conjunction with each other to dissect the molecular functions of each virulent factor in a cell-type-specific manner.