Effects of CXCL8 Overexpression on Tumor Cell Proliferation and Migration in an HNSCC Cell Model
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature
International Journal of Molecular Sciences Review The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature Jan Korbecki 1 , Klaudyna Kojder 2, Patrycja Kapczuk 1, Patrycja Kupnicka 1 , Barbara Gawro ´nska-Szklarz 3 , Izabela Gutowska 4 , Dariusz Chlubek 1 and Irena Baranowska-Bosiacka 1,* 1 Department of Biochemistry and Medical Chemistry, Pomeranian Medical University in Szczecin, Powsta´nców Wielkopolskich 72 Av., 70-111 Szczecin, Poland; [email protected] (J.K.); [email protected] (P.K.); [email protected] (P.K.); [email protected] (D.C.) 2 Department of Anaesthesiology and Intensive Care, Pomeranian Medical University in Szczecin, Unii Lubelskiej 1, 71-281 Szczecin, Poland; [email protected] 3 Department of Pharmacokinetics and Therapeutic Drug Monitoring, Pomeranian Medical University in Szczecin, Powsta´nców Wielkopolskich 72 Av., 70-111 Szczecin, Poland; [email protected] 4 Department of Medical Chemistry, Pomeranian Medical University in Szczecin, Powsta´nców Wlkp. 72 Av., 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-914661515 Abstract: Hypoxia is an integral component of the tumor microenvironment. Either as chronic or cycling hypoxia, it exerts a similar effect on cancer processes by activating hypoxia-inducible factor-1 (HIF-1) and nuclear factor (NF-κB), with cycling hypoxia showing a stronger proinflammatory influ- ence. One of the systems affected by hypoxia is the CXC chemokine system. This paper reviews all available information on hypoxia-induced changes in the expression of all CXC chemokines (CXCL1, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8 (IL-8), CXCL9, CXCL10, CXCL11, CXCL12 Citation: Korbecki, J.; Kojder, K.; Kapczuk, P.; Kupnicka, P.; (SDF-1), CXCL13, CXCL14, CXCL15, CXCL16, CXCL17) as well as CXC chemokine receptors— Gawro´nska-Szklarz,B.; Gutowska, I.; CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7 and CXCR8. -

Mouse CXCL15 ELISA Kit (ARG82062)
Product datasheet [email protected] ARG82062 Package: 96 wells Mouse CXCL15 ELISA Kit Store at: 4°C Summary Product Description Mouse CXCL15 ELISA Kit is an Enzyme Immunoassay kit for the quantification of Mouse CXCL15 in serum, plasma and cell culture supernatants. Tested Reactivity Ms Tested Application ELISA Target Name CXCL15 Conjugation HRP Conjugation Note Substrate: TMB and read at 450 nm. Sensitivity 62.5 pg/ml Sample Type Serum, plasma and cell culture supernatants. Standard Range 125 - 8000 pg/ml Sample Volume 100 µl Alternate Names Scyb15; weche; Small-inducible cytokine B15; C-X-C motif chemokine 15; Lungkine; lungkine Application Instructions Assay Time 4.5 hours Properties Form 96 well Storage instruction Store the kit at 2-8°C. Keep microplate wells sealed in a dry bag with desiccants. Do not expose test reagents to heat, sun or strong light during storage and usage. Please refer to the product user manual for detail temperatures of the components. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol Cxcl15 Gene Full Name chemokine (C-X-C motif) ligand 15 Function Chemotactic for neutrophils. Involved in lung-specific neutrophil trafficking during normal and inflammatory conditions. [UniProt] Highlight Related products: CXCL antibodies; CXCL ELISA Kits; CXCL Duos / Panels; CXCL recombinant proteins; New ELISA data calculation tool: Simplify the ELISA analysis by GainData www.arigobio.com 1/2 Images ARG82062 Mouse CXCL15 ELISA Kit standard curve image ARG82062 Mouse CXCL15 ELISA Kit results of a typical standard run with optical density reading at 450 nm. www.arigobio.com 2/2 Powered by TCPDF (www.tcpdf.org). -

The Chemokine System in Innate Immunity
Downloaded from http://cshperspectives.cshlp.org/ on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press The Chemokine System in Innate Immunity Caroline L. Sokol and Andrew D. Luster Center for Immunology & Inflammatory Diseases, Division of Rheumatology, Allergy and Immunology, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02114 Correspondence: [email protected] Chemokines are chemotactic cytokines that control the migration and positioning of immune cells in tissues and are critical for the function of the innate immune system. Chemokines control the release of innate immune cells from the bone marrow during homeostasis as well as in response to infection and inflammation. Theyalso recruit innate immune effectors out of the circulation and into the tissue where, in collaboration with other chemoattractants, they guide these cells to the very sites of tissue injury. Chemokine function is also critical for the positioning of innate immune sentinels in peripheral tissue and then, following innate immune activation, guiding these activated cells to the draining lymph node to initiate and imprint an adaptive immune response. In this review, we will highlight recent advances in understanding how chemokine function regulates the movement and positioning of innate immune cells at homeostasis and in response to acute inflammation, and then we will review how chemokine-mediated innate immune cell trafficking plays an essential role in linking the innate and adaptive immune responses. hemokines are chemotactic cytokines that with emphasis placed on its role in the innate Ccontrol cell migration and cell positioning immune system. throughout development, homeostasis, and in- flammation. The immune system, which is de- pendent on the coordinated migration of cells, CHEMOKINES AND CHEMOKINE RECEPTORS is particularly dependent on chemokines for its function. -

Chloride Channels Regulate Differentiation and Barrier Functions
RESEARCH ARTICLE Chloride channels regulate differentiation and barrier functions of the mammalian airway Mu He1†*, Bing Wu2†, Wenlei Ye1, Daniel D Le2, Adriane W Sinclair3,4, Valeria Padovano5, Yuzhang Chen6, Ke-Xin Li1, Rene Sit2, Michelle Tan2, Michael J Caplan5, Norma Neff2, Yuh Nung Jan1,7,8, Spyros Darmanis2*, Lily Yeh Jan1,7,8* 1Department of Physiology, University of California, San Francisco, San Francisco, United States; 2Chan Zuckerberg Biohub, San Francisco, United States; 3Department of Urology, University of California, San Francisco, San Francisco, United States; 4Division of Pediatric Urology, University of California, San Francisco, Benioff Children’s Hospital, San Francisco, United States; 5Department of Cellular and Molecular Physiology, Yale University School of Medicine, New Heaven, United States; 6Department of Anesthesia and Perioperative Care, University of California, San Francisco, San Francisco, United States; 7Department of Biochemistry and Biophysics, University of California, San Francisco, San Francisco, United States; 8Howard Hughes Medical Institute, University of California, San Francisco, San Francisco, United States *For correspondence: Abstract The conducting airway forms a protective mucosal barrier and is the primary target of [email protected] (MH); [email protected] airway disorders. The molecular events required for the formation and function of the airway (SD); mucosal barrier, as well as the mechanisms by which barrier dysfunction leads to early onset airway [email protected] (LYJ) diseases, -
Supplemental Figures 1-3 and Supplemental Table 1 (PDF, 155
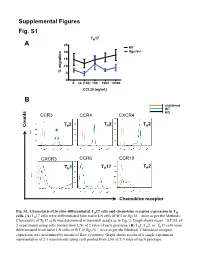
Supplemental Figures Fig. S1 TH17 A 25 WT 20 Rgs16-/- 15 10 % migration 5 0 0. ck (100) 100 1000 10000 CCL20 (ng/mL) B unstained WT KO CCR3 CCR4 CXCR4 T 2 T 2 T 2 Counts H H H CXCR3 CCR6 CCR10 T 2 TH1 TH17 H Chemokine receptor in vitro- Fig. S1. Chemotaxis of differentiated TH17 cells and chemokine receptor expression in TH –/– cells. (A) TH17 cells were differentiated from naïve LN cells of WT or Rgs16 mice as per the Methods. Chemotaxis of TH17 cells was determined in transwell assays as in Fig. 2. Graph shows mean ± S.E.M. of 2 experiments using cells pooled from LNs of 2 mice of each genotype. (B) TH1, TH2, or TH17 cells were differentiated from naïve LN cells of WT or Rgs16–/– mice as per the Methods. Chemokine receptor expression was determined by means of flow cytometry. Graph shows results of a single experiment representative of 2-3 experiments using cells pooled from LNs of 2-3 mice of each genotype. Fig. S2 A B 2.5 ) 6 2.0 1.5 1.0 Cells (x 10 0.5 0.0 WT Rgs16-/- Fig. S2. Intact proliferation and development RGS16-deficient TH cells. –/– (A) TH2 cells were differentiated from naïve LN cells of WT or Rgs16 mice as per the Methods. Proliferation of untreated (middle) or anti-CD3/ anti-CD28-treated (right) cells was determined by means of CFSE labeling –/– and flow cytometry. (B) Differentiated TH2 cells WT or Rgs16 mice were quantified after a 3 day incubation in basal medium. -

Recombinant Mouse CXCL15/Lungkine Catalog Number: 442-LK
Recombinant Mouse CXCL15/Lungkine Catalog Number: 442-LK DESCRIPTION Source E. coliderived Gln26Ala167, with an Nterminal Met Accession # Q9WVL7 Nterminal Sequence Met Analysis Predicted Molecular 16.5 kDa Mass SPECIFICATIONS Activity Bioassay data are not available. Endotoxin Level <0.01 EU per 1 μg of the protein by the LAL method. Purity >90%, by SDSPAGE under reducing conditions and visualized by silver stain. Formulation Lyophilized from a 0.2 μm filtered solution in Acetonitrile and TFA with BSA as a carrier protein. See Certificate of Analysis for details. PREPARATION AND STORAGE Reconstitution Reconstitute at 10 μg/mL in sterile PBS containing at least 0.1% human or bovine serum albumin. Shipping The product is shipped at ambient temperature. Upon receipt, store it immediately at the temperature recommended below. Stability & Storage Use a manual defrost freezer and avoid repeated freezethaw cycles. l 12 months from date of receipt, 20 to 70 °C as supplied. l 1 month, 2 to 8 °C under sterile conditions after reconstitution. l 3 months, 20 to 70 °C under sterile conditions after reconstitution. BACKGROUND Mouse Lungkine/CXCL15 (1), also named WECHE (2), is a member of the ELR motifcontaining CXC chemokines. The cDNA of mouse Lungkine encodes a protein of 166 amino acids (aa) with a 25 aa predicted signal peptide and a 141 aa mature protein with an extremely long C terminal tail that protrudes beyond the chemokine fold. Mouse Lungkine shares 35% aa sequence identity with human ENA78 and 31% identity with human IL8. The gene for mouse Lungkine has been mapped to chromosome 5. -

1 LETTER Castration-Mediated IL-8 Promotes Myeloid Infiltration And
bioRxiv preprint doi: https://doi.org/10.1101/651083; this version posted May 28, 2019. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. 1 LETTER 2 Castration-mediated IL-8 Promotes Myeloid Infiltration and Prostate Cancer 3 Progression 4 Keywords: androgen receptor, CXCL8, CXCR2, immunotherapy, PMN-MDSCs, 5 myeloid-derived suppresor cells. 1,2,3 1 3 6 Zoila A. Lopez-Bujanda , Michael C. Haffner , Matthew G. Chaimowitz , Nivedita 1,2,3 3 3 4 7 Chowdhury , Nicholas J. Venturini , Aleksandar Obradovic , Corey S. Hansen , 4 5 8 Joanna Jacków , Karen S. Sfanos1,5, Charles J. Bieberich6,7, Paula J. Hurley , Mark J. 8 8,9 4,10 9 Selby , Alan J. Korman , Angela M. Christiano , Angelo M. De Marzo1,5, and Charles 3,11,12 10 G. Drake . 11 1Department of Pathology, Johns Hopkins University School of Medicine, Baltimore, MD, 2 12 USA; Bloomberg-Kimmel Institute for Cancer Immunotherapy, Johns Hopkins University, 13 Baltimore, MD, USA; 3Columbia Center for Translational Immunology, Columbia 14 University Medical Center, New York, NY, USA; 4Department of Dermatology, Columbia 15 University, New York, NY, USA; 5Departments of Urology and Oncology, Johns Hopkins 16 University School of Medicine, Baltimore, MD, USA; 6Department of Biological Sciences, 17 University of Maryland Baltimore County, Baltimore, Maryland; 7University of Maryland 18 Marlene and Stewart Greenebaum Cancer Center, Baltimore, Maryland; 8Bristol-Myers 19 Squibb, Redwood City, CA, USA; 9Current affiliation: Vir Biotechnology, San Francisco, 10 20 CA; Department of Genetics & Development, Columbia University, New York, NY, USA; 1 bioRxiv preprint doi: https://doi.org/10.1101/651083; this version posted May 28, 2019. -

COMPREHENSIVE INVITED REVIEW Chemokines and Their Receptors
COMPREHENSIVE INVITED REVIEW Chemokines and Their Receptors Are Key Players in the Orchestra That Regulates Wound Healing Manuela Martins-Green,* Melissa Petreaca, and Lei Wang Department of Cell Biology and Neuroscience, University of California, Riverside, California. Significance: Normal wound healing progresses through a series of over- lapping phases, all of which are coordinated and regulated by a variety of molecules, including chemokines. Because these regulatory molecules play roles during the various stages of healing, alterations in their presence or function can lead to dysregulation of the wound-healing process, potentially leading to the development of chronic, nonhealing wounds. Recent Advances: A discovery that chemokines participate in a variety of disease conditions has propelled the study of these proteins to a level that potentially could lead to new avenues to treat disease. Their small size, ex- posed termini, and the fact that their only modifications are two disulfide Manuela Martins-Green, PhD bonds make them excellent targets for manipulation. In addition, because they bind to G-protein-coupled receptors (GPCRs), they are highly amenable to Submitted for publication January 9, 2013. *Correspondence: Department of Cell Biology pharmacological modulation. and Neuroscience, University of California, Riv- Critical Issues: Chemokines are multifunctional, and in many situations, their erside, Biological Sciences Building, 900 Uni- functions are highly dependent on the microenvironment. Moreover, each versity Ave., Riverside, CA 92521 (email: [email protected]). specific chemokine can bind to several GPCRs to stimulate the function, and both can function as monomers, homodimers, heterodimers, and even oligo- mers. Activation of one receptor by any single chemokine can lead to desen- Abbreviations sitization of other chemokine receptors, or even other GPCRs in the same cell, and Acronyms with implications for how these proteins or their receptors could be used to Ang-2 = angiopoietin-2 manipulate function. -

14,15-EET Induces the Infiltration and Tumor-Promoting Function of Neutrophils to Trigger the Growth of Minimal Dormant Metastases
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 28 Research Paper 14,15-EET induces the infiltration and tumor-promoting function of neutrophils to trigger the growth of minimal dormant metastases Jing Luo1,*, Xin-Xia Feng2,*, Chao Luo1, Yu Wang1, Dong Li1, Yu Shu1, Shan-Shan Wang1, Jian Qin1, Yong-Chao Li1, Jiu-Ming Zou1, De-An Tian2, Gui-Mei Zhang1, Zuo-Hua Feng1 1Department of Biochemistry and Molecular Biology, Wuhan University, School of Basic Medicine, Hubei, Wuhan 430030, People’s Republic of China 2Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Hongshan, Wuhan 430030, People’s Republic of China *These authors contributed equally to this work Correspondence to: Zuo-Hua Feng, email: [email protected] Gui-Mei Zhang, email: [email protected] Keywords: neutrophils, recruitment, metastases, dormancy, epoxyeicosatrienoic acids Received: January 06, 2016 Accepted: May 13, 2016 Published: May 30, 2016 ABSTRACT Infiltrating neutrophils are known to promote in the development of tumor. However, it is unclear whether and how neutrophils are involved in triggering the growth of dormant metastases. Here we show that 14,15-epoxyeicosatrienoic acid (14,15-EET) can trigger the growth of dormant micrometastases by inducing neutrophilic infiltration and converting neutrophil function. 14,15-EET triggered neutrophil infiltration in metastatic lesions by activating STAT3 and JNK pathways to induce the expression of human IL-8 and murine CXCL15 in corresponding tumor cells. The continuous expression of hIL-8/mCXCL15 was maintained by the sustained and enhanced activation of JNK pathway. 14,15-EET up-regulated miR-155 expression by activating STAT3 and JNK pathways. -

Products for Chemokine Research
RnDSy-lu-2945 Products for Chemokine Research The Chemokine Superfamily Chemokines are small cell surface-localized or secreted chemotactic cytokines that bind to and activate specific G protein-coupled chemokine receptors. Most chemokines have at least four conserved N-terminal cysteine residues that form two intramolecular disulfide bonds. Four chemokine subfamilies (CXC, CC, C and CX3C) have been defined based upon the placement of the first two cysteine residues. The CXC chemokine subfamily is characterized by two cysteine residues separated by one amino acid. Within this subfamily, two CXC classes are further defined by the presence or absence of an ELR motif sequence. ELR– CXC chemokines act as chemoattractants for lymphocytes, while ELR+ CXC chemokines are chemoattractants for neutrophils. Additionally, CXC chemokines can mediate angiogenesis.1 The CC chemokine subfamily is defined by two adjacent cysteine CCR1 H: CCL3-5, 7, 8, 13-16, 23, CCL3L1, CCL3L3, CCL4L1, CCL4L2 M: CCL3-7, 9/10 residues. CC chemokines induce inflammatory H: XCL1, 2 XCR1 responses via regulation of monocyte, M: XCL1 macrophage, mast cell, and T cell migration.2 H: CCL2, 7, 8, 13, 16 CCR2 (A or B) M: CCL2, 7, 12 C chemokines are characterized by a single H: CCL26 CX3CR1 cysteine residue and are constitutively expressed H: CX3CL1 in the thymus where they regulate T cell H: CCL5, 7, 8, 11, 13, 14, 15, 24, 26, 28, CCL3L1, CCL3L3 M: CX3CL1 CCR3 M: CCL5, 7, 9/10, 11, 24 differentiation.3 The CX3C chemokine subfamily is defined by two cysteine residues separated by H: CXCL6-8 CXCR1 three amino acids. -

Mouse and Human Immunology of Mice and Not Men: Differences
Of Mice and Not Men: Differences between Mouse and Human Immunology Javier Mestas and Christopher C. W. Hughes This information is current as J Immunol 2004; 172:2731-2738; ; of September 25, 2021. doi: 10.4049/jimmunol.172.5.2731 http://www.jimmunol.org/content/172/5/2731 Downloaded from References This article cites 101 articles, 27 of which you can access for free at: http://www.jimmunol.org/content/172/5/2731.full#ref-list-1 Why The JI? Submit online. http://www.jimmunol.org/ • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication *average by guest on September 25, 2021 Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2004 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. THE JOURNAL OF IMMUNOLOGY BRIEF REVIEWS Of Mice and Not Men: Differences between Mouse and Human Immunology Javier Mestas and Christopher C. W. Hughes1 Mice are the experimental tool of choice for the majority of sarily true in humans. By making such assumptions we run the immunologists and the study of their immune responses risk of overlooking aspects of human immunology that do not has yielded tremendous insight into the workings of the occur, or cannot be modeled, in mice. -

Lungkine (CXCL15), Murine Recombinant
BioVision 05/18 For research use only Lungkine (CXCL15), Murine Recombinant CATALOG NO: P1296-5, 20 5 µg, 20 µg RELATED PRODUCTS: ALTERNATE NAMES: CXCL15, Lungkine, Small-inducible cytokine B15, Scyb15, Weche, CXCL4, Human Recombinant (Cat. No. 7346) C-X-C motif chemokine 15 CXCL2, Human Recombinant (Cat. No. 7306) SOURCE: E.coli CXCL10/IP-10/CRG-2, human recombinant (Cat. No. 4277) CXCL14/BRAK, human recombinant (Cat. No.4278) PURITY: ≥ 98% by SDS-PAGE analyses CXCL16, Human Recombinant (Cat. No. 7130) MOL. WEIGHT: 16.3 kDa CXCL17, Human Recombinant (Cat. No. 7345) FORM: Lyophilized GCP-2 (CXCL6), human recombinant (Cat. No. 7153) IP-10, human recombinant (Cat. No. 4200) STORAGE CONDITIONS: Store at -20°C. After reconstitution, aliquot and store at -20°C to - MIG/CXCL9, murine recombinant (Cat. No. 7172) 80°C. Avoid repeated freezing and thawing cycles. PF-4/CXCL4, murine recombinant (Cat. No. 7187) BIOLOGICAL ACTIVITY: Determined by its ability to chemoattract human neutrophils using SDF-1beta (CXCL12), murine recombinant (Cat. No. 4391) a concentration range of 20.0-100.0 ng/ml SDF-1β/CXCL2, rat recombinant (Cat. No. 7200) RECONSTITUTION: Centrifuge the vial prior to opening. Reconstitute in water to a concentration of 0.1-1.0 mg/ml. Do not vortex. This solution can be stored at 2-8°C for up to 1 week. For extended storage, it is recommended reconstitute with sterilized distilled water or 0.1% BSA aqueous buffer and store in working aliquots at -20°C to - 80°C. DESCRIPTION: Lungkine is a CXC chemokine that is expressed in lung epithelial cells and, to a lesser extent, in certain fetal tissues.