Targeting the IL33–NLRP3 Axis Improves Therapy for Experimental Cerebral Malaria
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Integrated Analysis of Multiple Microarray Studies to Identify Novel Gene Signatures in Ulcerative Colitis
fgene-12-697514 July 5, 2021 Time: 19:1 # 1 ORIGINAL RESEARCH published: 09 July 2021 doi: 10.3389/fgene.2021.697514 Integrated Analysis of Multiple Microarray Studies to Identify Novel Gene Signatures in Ulcerative Colitis Zi-An Chen1, Yu-Feng Sun1, Quan-Xu Wang1, Hui-Hui Ma1, Zhi-Zhao Ma2* and Chuan-Jie Yang1* 1 Department of Gastroenterology, The Second Hospital of Hebei Medical University, Shijiazhuang, China, 2 Department of Neurosurgery, The Second Hospital of Hebei Medical University, Shijiazhuang, China Background: Ulcerative colitis (UC) is a chronic, complicated, inflammatory disease with an increasing incidence and prevalence worldwide. However, the intrinsic molecular mechanisms underlying the pathogenesis of UC have not yet been fully elucidated. Methods: All UC datasets published in the GEO database were analyzed and Edited by: Shulan Tian, summarized. Subsequently, the robust rank aggregation (RRA) method was used to Mayo Clinic, United States identify differentially expressed genes (DEGs) between UC patients and controls. Gene Reviewed by: functional annotation and PPI network analysis were performed to illustrate the potential Espiridión Ramos-Martínez, Universidad Nacional Autónoma functions of the DEGs. Some important functional modules from the protein-protein de México, Mexico interaction (PPI) network were identified by molecular complex detection (MCODE), Panwen Wang, Gene Ontology (GO), and Kyoto Encyclopedia of Genes and Genomes (KEGG), and Mayo Clinic Arizona, United States analyses were performed. The results of CytoHubba, a plug for integrated algorithm for *Correspondence: Zhi-Zhao Ma biomolecular interaction networks combined with RRA analysis, were used to identify [email protected] the hub genes. Finally, a mouse model of UC was established by dextran sulfate sodium Chuan-Jie Yang [email protected] salt (DSS) solution to verify the expression of hub genes. -

REG Gene Expression in Inflamed and Healthy Colon Mucosa Explored by in Situ Hybridisation
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Crossref Cell Tissue Res (2013) 352:639–646 DOI 10.1007/s00441-013-1592-z REGULAR ARTICLE REG gene expression in inflamed and healthy colon mucosa explored by in situ hybridisation Atle van Beelen Granlund & Ann Elisabet Østvik & Øystein Brenna & Sverre H. Torp & Bjørn I. Gustafsson & Arne Kristian Sandvik Received: 10 December 2012 /Accepted: 14 February 2013 /Published online: 22 March 2013 # The Author(s) 2013. This article is published with open access at Springerlink.com Abstract The regenerating islet-derived (REG) gene family used in situ hybridisation to demonstrate the cellular encodes a group of proteins highly expressed in several localisation of REG expression in healthy and diseased human pathologies, many of which are associated with colonic mucosa. Samples drawn from an IBD cohort includ- epithelial inflammation. All human family members, name- ing both inflamed and un-inflamed colonic mucosa are ly REG1A, REG1B, REG3A and REG4, are closely related described, as are samples from non-IBD inflammation and in genomic sequence and all are part of the c-type lectin healthy controls. Immunohistochemistry against known superfamily. REGs are highly expressed during inflamma- cell-type markers on serial sections has localised the expres- tory bowel disease (IBD)-related colonic inflammation and sion of REGs to metaplastic Paneth cells (REG1A, REG1B the in vivo expression pattern of REG1A and REG4 has and REG3A) and enteroendocrine cells (REG4), with a been localised by using immunohistochemistry. However, marked expansion of expression during inflammation. The the function of the encoded proteins is largely unknown and group of REGs can, based on gene expression patterns, be the cellular localisation of REG expression during colonic divided into at least two groups; REG1A, REG1B and inflammation has not been described. -

Identification of Novel Alternative Splice Isoforms of Circulating Proteins in a Mouse Model of Human Pancreatic Cancer
Research Article Identification of Novel Alternative Splice Isoforms of Circulating Proteins in a Mouse Model of Human Pancreatic Cancer Rajasree Menon,1 Qing Zhang,3 Yan Zhang,1 Damian Fermin,1 Nabeel Bardeesy,4 Ronald A. DePinho,5 Chunxia Lu,2 Samir M. Hanash,3 Gilbert S. Omenn,1 and David J. States1 1Center for Computational Medicine and Biology and 2Pediatric Endocrinology, University of Michigan, Ann Arbor, Michigan; 3Fred Hutchinson Cancer Research Center, Seattle, Washington; and 4Center for Cancer Research, Massachusetts General Hospital; 5Center for Applied Cancer Science, Dana-Farber Cancer Institute and Harvard Medical School, Boston, Massachusetts Abstract database are scored as high, medium, or low confidence, reflecting the amount of cumulative evidence in support of the existence of a To assess the potential of tumor-associated, alternatively particular alternatively spliced sequence. Evidence is collected from spliced gene products as a source of biomarkers in biological clustering of ESTs, mRNA sequences, and gene model predictions. fluids, we have analyzed a large data set of mass spectra We modified the ECgene database to include three-frame trans- derived from the plasma proteome of a mouse model of lations of the cDNA sequences (5) to determine the occurrence of human pancreatic ductal adenocarcinoma. MS/MS spectra novel splice variant proteins. An important development in recent were interrogated for novel splice isoforms using a non- years is the substantial improvement in tandem mass spectrometry redundant database containing an exhaustive three-frame instrumentation for proteomics, allowing in-depth analysis and translation of Ensembl transcripts and gene models from confident identifications even for proteins coded by mRNA ECgene. -

Investigation of the Underlying Hub Genes and Molexular Pathogensis in Gastric Cancer by Integrated Bioinformatic Analyses
bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Investigation of the underlying hub genes and molexular pathogensis in gastric cancer by integrated bioinformatic analyses Basavaraj Vastrad1, Chanabasayya Vastrad*2 1. Department of Biochemistry, Basaveshwar College of Pharmacy, Gadag, Karnataka 582103, India. 2. Biostatistics and Bioinformatics, Chanabasava Nilaya, Bharthinagar, Dharwad 580001, Karanataka, India. * Chanabasayya Vastrad [email protected] Ph: +919480073398 Chanabasava Nilaya, Bharthinagar, Dharwad 580001 , Karanataka, India bioRxiv preprint doi: https://doi.org/10.1101/2020.12.20.423656; this version posted December 22, 2020. The copyright holder for this preprint (which was not certified by peer review) is the author/funder. All rights reserved. No reuse allowed without permission. Abstract The high mortality rate of gastric cancer (GC) is in part due to the absence of initial disclosure of its biomarkers. The recognition of important genes associated in GC is therefore recommended to advance clinical prognosis, diagnosis and and treatment outcomes. The current investigation used the microarray dataset GSE113255 RNA seq data from the Gene Expression Omnibus database to diagnose differentially expressed genes (DEGs). Pathway and gene ontology enrichment analyses were performed, and a proteinprotein interaction network, modules, target genes - miRNA regulatory network and target genes - TF regulatory network were constructed and analyzed. Finally, validation of hub genes was performed. The 1008 DEGs identified consisted of 505 up regulated genes and 503 down regulated genes. -

The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature
International Journal of Molecular Sciences Review The Effect of Hypoxia on the Expression of CXC Chemokines and CXC Chemokine Receptors—A Review of Literature Jan Korbecki 1 , Klaudyna Kojder 2, Patrycja Kapczuk 1, Patrycja Kupnicka 1 , Barbara Gawro ´nska-Szklarz 3 , Izabela Gutowska 4 , Dariusz Chlubek 1 and Irena Baranowska-Bosiacka 1,* 1 Department of Biochemistry and Medical Chemistry, Pomeranian Medical University in Szczecin, Powsta´nców Wielkopolskich 72 Av., 70-111 Szczecin, Poland; [email protected] (J.K.); [email protected] (P.K.); [email protected] (P.K.); [email protected] (D.C.) 2 Department of Anaesthesiology and Intensive Care, Pomeranian Medical University in Szczecin, Unii Lubelskiej 1, 71-281 Szczecin, Poland; [email protected] 3 Department of Pharmacokinetics and Therapeutic Drug Monitoring, Pomeranian Medical University in Szczecin, Powsta´nców Wielkopolskich 72 Av., 70-111 Szczecin, Poland; [email protected] 4 Department of Medical Chemistry, Pomeranian Medical University in Szczecin, Powsta´nców Wlkp. 72 Av., 70-111 Szczecin, Poland; [email protected] * Correspondence: [email protected]; Tel.: +48-914661515 Abstract: Hypoxia is an integral component of the tumor microenvironment. Either as chronic or cycling hypoxia, it exerts a similar effect on cancer processes by activating hypoxia-inducible factor-1 (HIF-1) and nuclear factor (NF-κB), with cycling hypoxia showing a stronger proinflammatory influ- ence. One of the systems affected by hypoxia is the CXC chemokine system. This paper reviews all available information on hypoxia-induced changes in the expression of all CXC chemokines (CXCL1, CXCL2, CXCL3, CXCL4, CXCL5, CXCL6, CXCL7, CXCL8 (IL-8), CXCL9, CXCL10, CXCL11, CXCL12 Citation: Korbecki, J.; Kojder, K.; Kapczuk, P.; Kupnicka, P.; (SDF-1), CXCL13, CXCL14, CXCL15, CXCL16, CXCL17) as well as CXC chemokine receptors— Gawro´nska-Szklarz,B.; Gutowska, I.; CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, CXCR6, CXCR7 and CXCR8. -

Mouse CXCL15 ELISA Kit (ARG82062)
Product datasheet [email protected] ARG82062 Package: 96 wells Mouse CXCL15 ELISA Kit Store at: 4°C Summary Product Description Mouse CXCL15 ELISA Kit is an Enzyme Immunoassay kit for the quantification of Mouse CXCL15 in serum, plasma and cell culture supernatants. Tested Reactivity Ms Tested Application ELISA Target Name CXCL15 Conjugation HRP Conjugation Note Substrate: TMB and read at 450 nm. Sensitivity 62.5 pg/ml Sample Type Serum, plasma and cell culture supernatants. Standard Range 125 - 8000 pg/ml Sample Volume 100 µl Alternate Names Scyb15; weche; Small-inducible cytokine B15; C-X-C motif chemokine 15; Lungkine; lungkine Application Instructions Assay Time 4.5 hours Properties Form 96 well Storage instruction Store the kit at 2-8°C. Keep microplate wells sealed in a dry bag with desiccants. Do not expose test reagents to heat, sun or strong light during storage and usage. Please refer to the product user manual for detail temperatures of the components. Note For laboratory research only, not for drug, diagnostic or other use. Bioinformation Gene Symbol Cxcl15 Gene Full Name chemokine (C-X-C motif) ligand 15 Function Chemotactic for neutrophils. Involved in lung-specific neutrophil trafficking during normal and inflammatory conditions. [UniProt] Highlight Related products: CXCL antibodies; CXCL ELISA Kits; CXCL Duos / Panels; CXCL recombinant proteins; New ELISA data calculation tool: Simplify the ELISA analysis by GainData www.arigobio.com 1/2 Images ARG82062 Mouse CXCL15 ELISA Kit standard curve image ARG82062 Mouse CXCL15 ELISA Kit results of a typical standard run with optical density reading at 450 nm. www.arigobio.com 2/2 Powered by TCPDF (www.tcpdf.org). -

The Chemokine System in Innate Immunity
Downloaded from http://cshperspectives.cshlp.org/ on September 28, 2021 - Published by Cold Spring Harbor Laboratory Press The Chemokine System in Innate Immunity Caroline L. Sokol and Andrew D. Luster Center for Immunology & Inflammatory Diseases, Division of Rheumatology, Allergy and Immunology, Massachusetts General Hospital, Harvard Medical School, Boston, Massachusetts 02114 Correspondence: [email protected] Chemokines are chemotactic cytokines that control the migration and positioning of immune cells in tissues and are critical for the function of the innate immune system. Chemokines control the release of innate immune cells from the bone marrow during homeostasis as well as in response to infection and inflammation. Theyalso recruit innate immune effectors out of the circulation and into the tissue where, in collaboration with other chemoattractants, they guide these cells to the very sites of tissue injury. Chemokine function is also critical for the positioning of innate immune sentinels in peripheral tissue and then, following innate immune activation, guiding these activated cells to the draining lymph node to initiate and imprint an adaptive immune response. In this review, we will highlight recent advances in understanding how chemokine function regulates the movement and positioning of innate immune cells at homeostasis and in response to acute inflammation, and then we will review how chemokine-mediated innate immune cell trafficking plays an essential role in linking the innate and adaptive immune responses. hemokines are chemotactic cytokines that with emphasis placed on its role in the innate Ccontrol cell migration and cell positioning immune system. throughout development, homeostasis, and in- flammation. The immune system, which is de- pendent on the coordinated migration of cells, CHEMOKINES AND CHEMOKINE RECEPTORS is particularly dependent on chemokines for its function. -

Datasheet Blank Template
SAN TA C RUZ BI OTEC HNOL OG Y, INC . Reg lll α/γ (S-13): sc-103862 BACKGROUND APPLICATIONS The regeneration (Reg) family consists of secretory proteins involved in liver, Reg III α/γ (S-13) is recommended for detection of Reg III α and Reg III γ of pancreatic, gastric and intestinal cell proliferation or differentiation. Members human origin by Western Blotting (starting dilution 1:200, dilution range of the Reg family are divided into four subclasses, designated types I, II, III 1:100-1:1000), immunoprecipitation [1-2 µg per 100-500 µg of total protein and IV, all of which share a common gene structure containing five introns and (1 ml of cell lysate)], immunofluorescence (starting dilution 1:50, dilution six exons. Members of the Reg family have been implicated in the regulation range 1:50-1:500) and solid phase ELISA (starting dilution 1:30, dilution of cell growth, tumorigenesis and the progression of cancer. Reg lll γ (regen - range 1:30-1:3000). erating islet-derived 3 γ), also known as pancreatitis-associated protein 1B, PA P1B or UNQ429, is a 175 amino acid secreted protein that is expressed Molecular Weight of Reg III α: 19 kDa. almost exclusively in pancreas, with low levels of expression in testis. Reg lll γ Molecular Weight of Reg lll γ: 16 kDa. functions as an antimicrobial protein involved in controlling bacterial prolifera - Positive Controls: human pancreas extract: sc-363770. tion and may be induced during acute pancreatitis. The gene encoding Reg lll γ maps to human chromosome 2, which houses over 1,400 genes and comprises nearly 8% of the human genome. -

Reg Proteins Promote Acinar-To-Ductal Metaplasia and Act As Novel Diagnostic and Prognostic Markers in Pancreatic Ductal Adenocarcinoma
www.impactjournals.com/oncotarget/ Oncotarget, Vol. 7, No. 47 Research Paper Reg proteins promote acinar-to-ductal metaplasia and act as novel diagnostic and prognostic markers in pancreatic ductal adenocarcinoma Qing Li1,*, Hao Wang2,*, George Zogopoulos3,4, Qin Shao5, Kun Dong6, Fudong Lv6, Karam Nwilati1, Xian-yong Gui7, Adeline Cuggia4, Jun-Li Liu1, Zu-hua Gao5 1Fraser Laboratories for Diabetes Research, Department of Medicine, McGill University Health Centre, Montreal, QC, Canada 2Department of Oncology, Qingdao Municipal Hospital, School of Medicine, Qingdao University, Qingdao, China 3Department of Surgery, McGill University Health Centre, Montreal, QC, Canada 4Quebec Pancreas Cancer Study, McGill University Health Centre, Montreal, QC, Canada 5Department of Pathology, McGill University Health Centre, Montreal, QC, Canada 6Department of Pathology, You An Hospital, Capital Medical University, Beijing, China 7Department of Pathology, University of Calgary, Calgary, AB, Canada *These authors have contributed equally Correspondence to: Zu-hua Gao, email: [email protected] Jun-Li Liu, email: [email protected] Keywords: Reg family proteins, pancreatic ductal adenocarcinoma, acinar-to-ductal metaplasia, pancreatic intraepithelial neoplasia, cholangiocarcinoma Received: March 01, 2016 Accepted: October 13, 2016 Published: October 24, 2016 ABSTRACT Pancreatic ductal adenocarcinoma (PDAC) is a highly aggressive malignant tumor. Acinar-to-ductal metaplasia (ADM) and pancreatic intraepithelial neoplasia (PanIN) are both precursor lesions that lead to the development of PDAC. Reg family proteins (Reg1A, 1B, 3A/G, 4) are a group of calcium-dependent lectins that promote islet growth in response to inflammation and/or injuries. The aim of this study was to establish a role for Reg proteins in the development of PDAC and their clinical value as biomarkers. -

Chloride Channels Regulate Differentiation and Barrier Functions
RESEARCH ARTICLE Chloride channels regulate differentiation and barrier functions of the mammalian airway Mu He1†*, Bing Wu2†, Wenlei Ye1, Daniel D Le2, Adriane W Sinclair3,4, Valeria Padovano5, Yuzhang Chen6, Ke-Xin Li1, Rene Sit2, Michelle Tan2, Michael J Caplan5, Norma Neff2, Yuh Nung Jan1,7,8, Spyros Darmanis2*, Lily Yeh Jan1,7,8* 1Department of Physiology, University of California, San Francisco, San Francisco, United States; 2Chan Zuckerberg Biohub, San Francisco, United States; 3Department of Urology, University of California, San Francisco, San Francisco, United States; 4Division of Pediatric Urology, University of California, San Francisco, Benioff Children’s Hospital, San Francisco, United States; 5Department of Cellular and Molecular Physiology, Yale University School of Medicine, New Heaven, United States; 6Department of Anesthesia and Perioperative Care, University of California, San Francisco, San Francisco, United States; 7Department of Biochemistry and Biophysics, University of California, San Francisco, San Francisco, United States; 8Howard Hughes Medical Institute, University of California, San Francisco, San Francisco, United States *For correspondence: Abstract The conducting airway forms a protective mucosal barrier and is the primary target of [email protected] (MH); [email protected] airway disorders. The molecular events required for the formation and function of the airway (SD); mucosal barrier, as well as the mechanisms by which barrier dysfunction leads to early onset airway [email protected] (LYJ) diseases, -

Human Lectins, Their Carbohydrate Affinities and Where to Find Them
biomolecules Review Human Lectins, Their Carbohydrate Affinities and Where to Review HumanFind Them Lectins, Their Carbohydrate Affinities and Where to FindCláudia ThemD. Raposo 1,*, André B. Canelas 2 and M. Teresa Barros 1 1, 2 1 Cláudia D. Raposo * , Andr1 é LAQVB. Canelas‐Requimte,and Department M. Teresa of Chemistry, Barros NOVA School of Science and Technology, Universidade NOVA de Lisboa, 2829‐516 Caparica, Portugal; [email protected] 12 GlanbiaLAQV-Requimte,‐AgriChemWhey, Department Lisheen of Chemistry, Mine, Killoran, NOVA Moyne, School E41 of ScienceR622 Co. and Tipperary, Technology, Ireland; canelas‐ [email protected] NOVA de Lisboa, 2829-516 Caparica, Portugal; [email protected] 2* Correspondence:Glanbia-AgriChemWhey, [email protected]; Lisheen Mine, Tel.: Killoran, +351‐212948550 Moyne, E41 R622 Tipperary, Ireland; [email protected] * Correspondence: [email protected]; Tel.: +351-212948550 Abstract: Lectins are a class of proteins responsible for several biological roles such as cell‐cell in‐ Abstract:teractions,Lectins signaling are pathways, a class of and proteins several responsible innate immune for several responses biological against roles pathogens. such as Since cell-cell lec‐ interactions,tins are able signalingto bind to pathways, carbohydrates, and several they can innate be a immuneviable target responses for targeted against drug pathogens. delivery Since sys‐ lectinstems. In are fact, able several to bind lectins to carbohydrates, were approved they by canFood be and a viable Drug targetAdministration for targeted for drugthat purpose. delivery systems.Information In fact, about several specific lectins carbohydrate were approved recognition by Food by andlectin Drug receptors Administration was gathered for that herein, purpose. plus Informationthe specific organs about specific where those carbohydrate lectins can recognition be found by within lectin the receptors human was body. -
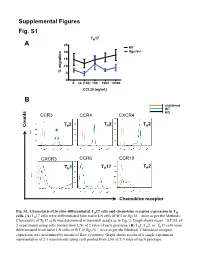
Supplemental Figures 1-3 and Supplemental Table 1 (PDF, 155
Supplemental Figures Fig. S1 TH17 A 25 WT 20 Rgs16-/- 15 10 % migration 5 0 0. ck (100) 100 1000 10000 CCL20 (ng/mL) B unstained WT KO CCR3 CCR4 CXCR4 T 2 T 2 T 2 Counts H H H CXCR3 CCR6 CCR10 T 2 TH1 TH17 H Chemokine receptor in vitro- Fig. S1. Chemotaxis of differentiated TH17 cells and chemokine receptor expression in TH –/– cells. (A) TH17 cells were differentiated from naïve LN cells of WT or Rgs16 mice as per the Methods. Chemotaxis of TH17 cells was determined in transwell assays as in Fig. 2. Graph shows mean ± S.E.M. of 2 experiments using cells pooled from LNs of 2 mice of each genotype. (B) TH1, TH2, or TH17 cells were differentiated from naïve LN cells of WT or Rgs16–/– mice as per the Methods. Chemokine receptor expression was determined by means of flow cytometry. Graph shows results of a single experiment representative of 2-3 experiments using cells pooled from LNs of 2-3 mice of each genotype. Fig. S2 A B 2.5 ) 6 2.0 1.5 1.0 Cells (x 10 0.5 0.0 WT Rgs16-/- Fig. S2. Intact proliferation and development RGS16-deficient TH cells. –/– (A) TH2 cells were differentiated from naïve LN cells of WT or Rgs16 mice as per the Methods. Proliferation of untreated (middle) or anti-CD3/ anti-CD28-treated (right) cells was determined by means of CFSE labeling –/– and flow cytometry. (B) Differentiated TH2 cells WT or Rgs16 mice were quantified after a 3 day incubation in basal medium.