Chloride Channels Regulate Differentiation and Barrier Functions
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Induces Homing of Antigen-Specific and Non Chemokine-Adjuvanted
Chemokine-Adjuvanted Plasmid DNA Induces Homing of Antigen-Specific and Non −Antigen-Specific B and T Cells to the Intestinal and Genital Mucosae This information is current as of September 25, 2021. Yoann Aldon, Sven Kratochvil, Robin J. Shattock and Paul F. McKay J Immunol published online 8 January 2020 http://www.jimmunol.org/content/early/2020/01/07/jimmun ol.1901184 Downloaded from Supplementary http://www.jimmunol.org/content/suppl/2020/01/07/jimmunol.190118 Material 4.DCSupplemental http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 25, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Author Choice Freely available online through The Journal of Immunology Author Choice option Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2020 The Authors All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. Published January 8, 2020, doi:10.4049/jimmunol.1901184 The Journal of Immunology Chemokine-Adjuvanted Plasmid DNA Induces Homing of Antigen-Specific and Non–Antigen-Specific B and T Cells to the Intestinal and Genital Mucosae Yoann Aldon, Sven Kratochvil, Robin J. -

Supplementary Materials: Evaluation of Cytotoxicity and Α-Glucosidase Inhibitory Activity of Amide and Polyamino-Derivatives of Lupane Triterpenoids
Supplementary Materials: Evaluation of cytotoxicity and α-glucosidase inhibitory activity of amide and polyamino-derivatives of lupane triterpenoids Oxana B. Kazakova1*, Gul'nara V. Giniyatullina1, Akhat G. Mustafin1, Denis A. Babkov2, Elena V. Sokolova2, Alexander A. Spasov2* 1Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences, 71, pr. Oktyabrya, 450054 Ufa, Russian Federation 2Scientific Center for Innovative Drugs, Volgograd State Medical University, Novorossiyskaya st. 39, Volgograd 400087, Russian Federation Correspondence Prof. Dr. Oxana B. Kazakova Ufa Institute of Chemistry of the Ufa Federal Research Centre of the Russian Academy of Sciences 71 Prospeсt Oktyabrya Ufa, 450054 Russian Federation E-mail: [email protected] Prof. Dr. Alexander A. Spasov Scientific Center for Innovative Drugs of the Volgograd State Medical University 39 Novorossiyskaya st. Volgograd, 400087 Russian Federation E-mail: [email protected] Figure S1. 1H and 13C of compound 2. H NH N H O H O H 2 2 Figure S2. 1H and 13C of compound 4. NH2 O H O H CH3 O O H H3C O H 4 3 Figure S3. Anticancer screening data of compound 2 at single dose assay 4 Figure S4. Anticancer screening data of compound 7 at single dose assay 5 Figure S5. Anticancer screening data of compound 8 at single dose assay 6 Figure S6. Anticancer screening data of compound 9 at single dose assay 7 Figure S7. Anticancer screening data of compound 12 at single dose assay 8 Figure S8. Anticancer screening data of compound 13 at single dose assay 9 Figure S9. Anticancer screening data of compound 14 at single dose assay 10 Figure S10. -

Prospective Isolation of NKX2-1–Expressing Human Lung Progenitors Derived from Pluripotent Stem Cells
The Journal of Clinical Investigation RESEARCH ARTICLE Prospective isolation of NKX2-1–expressing human lung progenitors derived from pluripotent stem cells Finn Hawkins,1,2 Philipp Kramer,3 Anjali Jacob,1,2 Ian Driver,4 Dylan C. Thomas,1 Katherine B. McCauley,1,2 Nicholas Skvir,1 Ana M. Crane,3 Anita A. Kurmann,1,5 Anthony N. Hollenberg,5 Sinead Nguyen,1 Brandon G. Wong,6 Ahmad S. Khalil,6,7 Sarah X.L. Huang,3,8 Susan Guttentag,9 Jason R. Rock,4 John M. Shannon,10 Brian R. Davis,3 and Darrell N. Kotton1,2 2 1Center for Regenerative Medicine, and The Pulmonary Center and Department of Medicine, Boston University School of Medicine, Boston, Massachusetts, USA. 3Center for Stem Cell and Regenerative Medicine, Brown Foundation Institute of Molecular Medicine, University of Texas Health Science Center, Houston, Texas, USA. 4Department of Anatomy, UCSF, San Francisco, California, USA. 5Division of Endocrinology, Diabetes and Metabolism, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, Massachusetts, USA. 6Department of Biomedical Engineering and Biological Design Center, Boston University, Boston, Massachusetts, USA. 7Wyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts, USA. 8Columbia Center for Translational Immunology & Columbia Center for Human Development, Columbia University Medical Center, New York, New York, USA. 9Department of Pediatrics, Monroe Carell Jr. Children’s Hospital, Vanderbilt University, Nashville, Tennessee, USA. 10Division of Pulmonary Biology, Cincinnati Children’s Hospital, Cincinnati, Ohio, USA. It has been postulated that during human fetal development, all cells of the lung epithelium derive from embryonic, endodermal, NK2 homeobox 1–expressing (NKX2-1+) precursor cells. -

Examination of the Transcription Factors Acting in Bone Marrow
THESIS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY (PHD) Examination of the transcription factors acting in bone marrow derived macrophages by Gergely Nagy Supervisor: Dr. Endre Barta UNIVERSITY OF DEBRECEN DOCTORAL SCHOOL OF MOLECULAR CELL AND IMMUNE BIOLOGY DEBRECEN, 2016 Table of contents Table of contents ........................................................................................................................ 2 1. Introduction ............................................................................................................................ 5 1.1. Transcriptional regulation ................................................................................................... 5 1.1.1. Transcriptional initiation .................................................................................................. 5 1.1.2. Co-regulators and histone modifications .......................................................................... 8 1.2. Promoter and enhancer sequences guiding transcription factors ...................................... 11 1.2.1. General transcription factors .......................................................................................... 11 1.2.2. The ETS superfamily ..................................................................................................... 17 1.2.3. The AP-1 and CREB proteins ........................................................................................ 20 1.2.4. Other promoter specific transcription factor families ................................................... -

Supplementary Information Integrative Analyses of Splicing in the Aging Brain: Role in Susceptibility to Alzheimer’S Disease
Supplementary Information Integrative analyses of splicing in the aging brain: role in susceptibility to Alzheimer’s Disease Contents 1. Supplementary Notes 1.1. Religious Orders Study and Memory and Aging Project 1.2. Mount Sinai Brain Bank Alzheimer’s Disease 1.3. CommonMind Consortium 1.4. Data Availability 2. Supplementary Tables 3. Supplementary Figures Note: Supplementary Tables are provided as separate Excel files. 1. Supplementary Notes 1.1. Religious Orders Study and Memory and Aging Project Gene expression data1. Gene expression data were generated using RNA- sequencing from Dorsolateral Prefrontal Cortex (DLPFC) of 540 individuals, at an average sequence depth of 90M reads. Detailed description of data generation and processing was previously described2 (Mostafavi, Gaiteri et al., under review). Samples were submitted to the Broad Institute’s Genomics Platform for transcriptome analysis following the dUTP protocol with Poly(A) selection developed by Levin and colleagues3. All samples were chosen to pass two initial quality filters: RNA integrity (RIN) score >5 and quantity threshold of 5 ug (and were selected from a larger set of 724 samples). Sequencing was performed on the Illumina HiSeq with 101bp paired-end reads and achieved coverage of 150M reads of the first 12 samples. These 12 samples will serve as a deep coverage reference and included 2 males and 2 females of nonimpaired, mild cognitive impaired, and Alzheimer's cases. The remaining samples were sequenced with target coverage of 50M reads; the mean coverage for the samples passing QC is 95 million reads (median 90 million reads). The libraries were constructed and pooled according to the RIN scores such that similar RIN scores would be pooled together. -

Human B Cells Expression in Terminally Differentiating Induces
1,25-Dihydroxyvitamin D3 Induces CCR10 Expression in Terminally Differentiating Human B Cells This information is current as Aiko-Konno Shirakawa, Daisuke Nagakubo, Kunio of September 28, 2021. Hieshima, Takashi Nakayama, Zhe Jin and Osamu Yoshie J Immunol 2008; 180:2786-2795; ; doi: 10.4049/jimmunol.180.5.2786 http://www.jimmunol.org/content/180/5/2786 Downloaded from References This article cites 55 articles, 29 of which you can access for free at: http://www.jimmunol.org/content/180/5/2786.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists • Fast Publication! 4 weeks from acceptance to publication by guest on September 28, 2021 *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2008 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology 1,25-Dihydroxyvitamin D3 Induces CCR10 Expression in Terminally Differentiating Human B Cells1 Aiko-Konno Shirakawa,2 Daisuke Nagakubo,2 Kunio Hieshima, Takashi Nakayama, Zhe Jin, and Osamu Yoshie3 In the B cell lineage, CCR10 is known to be selectively expressed by plasma cells, especially those secreting IgA. -

A Computational Approach for Defining a Signature of Β-Cell Golgi Stress in Diabetes Mellitus
Page 1 of 781 Diabetes A Computational Approach for Defining a Signature of β-Cell Golgi Stress in Diabetes Mellitus Robert N. Bone1,6,7, Olufunmilola Oyebamiji2, Sayali Talware2, Sharmila Selvaraj2, Preethi Krishnan3,6, Farooq Syed1,6,7, Huanmei Wu2, Carmella Evans-Molina 1,3,4,5,6,7,8* Departments of 1Pediatrics, 3Medicine, 4Anatomy, Cell Biology & Physiology, 5Biochemistry & Molecular Biology, the 6Center for Diabetes & Metabolic Diseases, and the 7Herman B. Wells Center for Pediatric Research, Indiana University School of Medicine, Indianapolis, IN 46202; 2Department of BioHealth Informatics, Indiana University-Purdue University Indianapolis, Indianapolis, IN, 46202; 8Roudebush VA Medical Center, Indianapolis, IN 46202. *Corresponding Author(s): Carmella Evans-Molina, MD, PhD ([email protected]) Indiana University School of Medicine, 635 Barnhill Drive, MS 2031A, Indianapolis, IN 46202, Telephone: (317) 274-4145, Fax (317) 274-4107 Running Title: Golgi Stress Response in Diabetes Word Count: 4358 Number of Figures: 6 Keywords: Golgi apparatus stress, Islets, β cell, Type 1 diabetes, Type 2 diabetes 1 Diabetes Publish Ahead of Print, published online August 20, 2020 Diabetes Page 2 of 781 ABSTRACT The Golgi apparatus (GA) is an important site of insulin processing and granule maturation, but whether GA organelle dysfunction and GA stress are present in the diabetic β-cell has not been tested. We utilized an informatics-based approach to develop a transcriptional signature of β-cell GA stress using existing RNA sequencing and microarray datasets generated using human islets from donors with diabetes and islets where type 1(T1D) and type 2 diabetes (T2D) had been modeled ex vivo. To narrow our results to GA-specific genes, we applied a filter set of 1,030 genes accepted as GA associated. -

Supplementary Table 3 Complete List of RNA-Sequencing Analysis of Gene Expression Changed by ≥ Tenfold Between Xenograft and Cells Cultured in 10%O2
Supplementary Table 3 Complete list of RNA-Sequencing analysis of gene expression changed by ≥ tenfold between xenograft and cells cultured in 10%O2 Expr Log2 Ratio Symbol Entrez Gene Name (culture/xenograft) -7.182 PGM5 phosphoglucomutase 5 -6.883 GPBAR1 G protein-coupled bile acid receptor 1 -6.683 CPVL carboxypeptidase, vitellogenic like -6.398 MTMR9LP myotubularin related protein 9-like, pseudogene -6.131 SCN7A sodium voltage-gated channel alpha subunit 7 -6.115 POPDC2 popeye domain containing 2 -6.014 LGI1 leucine rich glioma inactivated 1 -5.86 SCN1A sodium voltage-gated channel alpha subunit 1 -5.713 C6 complement C6 -5.365 ANGPTL1 angiopoietin like 1 -5.327 TNN tenascin N -5.228 DHRS2 dehydrogenase/reductase 2 leucine rich repeat and fibronectin type III domain -5.115 LRFN2 containing 2 -5.076 FOXO6 forkhead box O6 -5.035 ETNPPL ethanolamine-phosphate phospho-lyase -4.993 MYO15A myosin XVA -4.972 IGF1 insulin like growth factor 1 -4.956 DLG2 discs large MAGUK scaffold protein 2 -4.86 SCML4 sex comb on midleg like 4 (Drosophila) Src homology 2 domain containing transforming -4.816 SHD protein D -4.764 PLP1 proteolipid protein 1 -4.764 TSPAN32 tetraspanin 32 -4.713 N4BP3 NEDD4 binding protein 3 -4.705 MYOC myocilin -4.646 CLEC3B C-type lectin domain family 3 member B -4.646 C7 complement C7 -4.62 TGM2 transglutaminase 2 -4.562 COL9A1 collagen type IX alpha 1 chain -4.55 SOSTDC1 sclerostin domain containing 1 -4.55 OGN osteoglycin -4.505 DAPL1 death associated protein like 1 -4.491 C10orf105 chromosome 10 open reading frame 105 -4.491 -

Antimicrobial Activity As a Chemokine with Broad-Spectrum CCL28 Has
CCL28 Has Dual Roles in Mucosal Immunity as a Chemokine with Broad-Spectrum Antimicrobial Activity This information is current as Kunio Hieshima, Haruo Ohtani, Michiko Shibano, Dai of September 26, 2021. Izawa, Takashi Nakayama, Yuri Kawasaki, Fumio Shiba, Mitsuru Shiota, Fuminori Katou, Takuya Saito and Osamu Yoshie J Immunol 2003; 170:1452-1461; ; doi: 10.4049/jimmunol.170.3.1452 Downloaded from http://www.jimmunol.org/content/170/3/1452 References This article cites 36 articles, 19 of which you can access for free at: http://www.jimmunol.org/content/170/3/1452.full#ref-list-1 http://www.jimmunol.org/ Why The JI? Submit online. • Rapid Reviews! 30 days* from submission to initial decision • No Triage! Every submission reviewed by practicing scientists by guest on September 26, 2021 • Fast Publication! 4 weeks from acceptance to publication *average Subscription Information about subscribing to The Journal of Immunology is online at: http://jimmunol.org/subscription Permissions Submit copyright permission requests at: http://www.aai.org/About/Publications/JI/copyright.html Email Alerts Receive free email-alerts when new articles cite this article. Sign up at: http://jimmunol.org/alerts The Journal of Immunology is published twice each month by The American Association of Immunologists, Inc., 1451 Rockville Pike, Suite 650, Rockville, MD 20852 Copyright © 2003 by The American Association of Immunologists All rights reserved. Print ISSN: 0022-1767 Online ISSN: 1550-6606. The Journal of Immunology CCL28 Has Dual Roles in Mucosal Immunity as a Chemokine with Broad-Spectrum Antimicrobial Activity1 Kunio Hieshima,* Haruo Ohtani,2‡ Michiko Shibano,* Dai Izawa,* Takashi Nakayama,* Yuri Kawasaki,* Fumio Shiba,* Mitsuru Shiota,† Fuminori Katou,§ Takuya Saito,¶ and Osamu Yoshie3* CCL28 is a CC chemokine signaling via CCR10 and CCR3 that is selectively expressed in certain mucosal tissues such as exocrine glands, trachea, and colon. -

4-6 Weeks Old Female C57BL/6 Mice Obtained from Jackson Labs Were Used for Cell Isolation
Methods Mice: 4-6 weeks old female C57BL/6 mice obtained from Jackson labs were used for cell isolation. Female Foxp3-IRES-GFP reporter mice (1), backcrossed to B6/C57 background for 10 generations, were used for the isolation of naïve CD4 and naïve CD8 cells for the RNAseq experiments. The mice were housed in pathogen-free animal facility in the La Jolla Institute for Allergy and Immunology and were used according to protocols approved by the Institutional Animal Care and use Committee. Preparation of cells: Subsets of thymocytes were isolated by cell sorting as previously described (2), after cell surface staining using CD4 (GK1.5), CD8 (53-6.7), CD3ε (145- 2C11), CD24 (M1/69) (all from Biolegend). DP cells: CD4+CD8 int/hi; CD4 SP cells: CD4CD3 hi, CD24 int/lo; CD8 SP cells: CD8 int/hi CD4 CD3 hi, CD24 int/lo (Fig S2). Peripheral subsets were isolated after pooling spleen and lymph nodes. T cells were enriched by negative isolation using Dynabeads (Dynabeads untouched mouse T cells, 11413D, Invitrogen). After surface staining for CD4 (GK1.5), CD8 (53-6.7), CD62L (MEL-14), CD25 (PC61) and CD44 (IM7), naïve CD4+CD62L hiCD25-CD44lo and naïve CD8+CD62L hiCD25-CD44lo were obtained by sorting (BD FACS Aria). Additionally, for the RNAseq experiments, CD4 and CD8 naïve cells were isolated by sorting T cells from the Foxp3- IRES-GFP mice: CD4+CD62LhiCD25–CD44lo GFP(FOXP3)– and CD8+CD62LhiCD25– CD44lo GFP(FOXP3)– (antibodies were from Biolegend). In some cases, naïve CD4 cells were cultured in vitro under Th1 or Th2 polarizing conditions (3, 4). -

Molecular Pathways Associated with the Nutritional Programming of Plant-Based Diet Acceptance in Rainbow Trout Following an Early Feeding Exposure Mukundh N
Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, Aurélie Le Cam, Françoise Médale, Sadasivam J. Kaushik, Inge Geurden To cite this version: Mukundh N. Balasubramanian, Stéphane Panserat, Mathilde Dupont-Nivet, Edwige Quillet, Jérôme Montfort, et al.. Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure. BMC Genomics, BioMed Central, 2016, 17 (1), pp.1-20. 10.1186/s12864-016-2804-1. hal-01346912 HAL Id: hal-01346912 https://hal.archives-ouvertes.fr/hal-01346912 Submitted on 19 Jul 2016 HAL is a multi-disciplinary open access L’archive ouverte pluridisciplinaire HAL, est archive for the deposit and dissemination of sci- destinée au dépôt et à la diffusion de documents entific research documents, whether they are pub- scientifiques de niveau recherche, publiés ou non, lished or not. The documents may come from émanant des établissements d’enseignement et de teaching and research institutions in France or recherche français ou étrangers, des laboratoires abroad, or from public or private research centers. publics ou privés. Balasubramanian et al. BMC Genomics (2016) 17:449 DOI 10.1186/s12864-016-2804-1 RESEARCH ARTICLE Open Access Molecular pathways associated with the nutritional programming of plant-based diet acceptance in rainbow trout following an early feeding exposure Mukundh N. Balasubramanian1, Stephane Panserat1, Mathilde Dupont-Nivet2, Edwige Quillet2, Jerome Montfort3, Aurelie Le Cam3, Francoise Medale1, Sadasivam J. Kaushik1 and Inge Geurden1* Abstract Background: The achievement of sustainable feeding practices in aquaculture by reducing the reliance on wild-captured fish, via replacement of fish-based feed with plant-based feed, is impeded by the poor growth response seen in fish fed high levels of plant ingredients. -
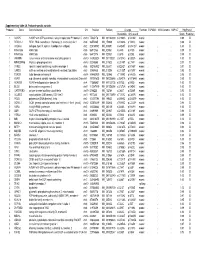
Supplementary Table 2A. Proband-Specific Variants Proband
Supplementary table 2A. Proband-specific variants Proband Gene Gene full name Chr Position RefSeqChange Function ESP6500 1000 Genome SNP137 PolyPhen2 Nucleotide Amino acid Score Prediction 1 AGAP5 ArfGAP with GTPase domain, ankyrin repeat and PH domain 5 chr10 75435178 NM_001144000 c.G1240A p.V414M exonic . 0.99 D 1 REXO1L1 REX1, RNA exonuclease 1 homolog (S. cerevisiae)-like 1 chr8 86574465 NM_172239 c.A1262G p.Y421C exonic . 0.98 D 1 COL4A3 collagen, type IV, alpha 3 (Goodpasture antigen) chr2 228169782 NM_000091 c.G4235T p.G1412V exonic . 1.00 D 1 KIAA1586 KIAA1586 chr6 56912133 NM_020931 c.C44G p.A15G exonic . 0.99 D 1 KIAA1586 KIAA1586 chr6 56912176 NM_020931 c.C87G p.D29E exonic . 0.99 D 1 JAKMIP3 Janus kinase and microtubule interacting protein 3 chr10 133955524 NM_001105521 c.G1574C p.G525A exonic . 1.00 D 1 MPHOSPH8 M-phase phosphoprotein 8 chr13 20240685 NM_017520 c.C2140T p.L714F exonic . 0.97 D 1 SYNE1 spectrin repeat containing, nuclear envelope 1 chr6 152783902 NM_033071 c.A2242T p.N748Y exonic . 0.97 D 1 CAND2 cullin-associated and neddylation-dissociated 2 (putative) chr3 12858835 NM_012298 c.C2125T p.H709Y exonic . 0.95 D 1 TDRD9 tudor domain containing 9 chr14 104460909 NM_153046 c.T1289G p.V430G exonic . 0.96 D 2 ASPM asp (abnormal spindle) homolog, microcephaly associated (Drosophila)chr1 197091625 NM_001206846 c.G3491A p.R1164H exonic . 0.99 D 2 ADAM29 ADAM metallopeptidase domain 29 chr4 175896951 NM_001130705 c.A275G p.D92G exonic . 1.00 D 2 BCO2 beta-carotene oxygenase 2 chr11 112087019 NM_001256398 c.G1373A p.G458E exonic .