Rearrangement Studies and Novel Reactions of Organocadmium Reagents
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

(A) 1) at First a Pair of Each 10G. of Sample Was Extracted with Ether by the Soxhlet's Apparatus for a Week
52 [vol. 6, THE DISTRIBUTION OF PHOSPHORUS IN COW'S MILK AND THE SCHEME FOR THE SEPARATION OF PHOSPHATIDES By Rinjiro SASAKI (From the Institute of Agricultural Chemistry, College of Agriculture, Tokyo Imperial University) (Received 9th September 1930) A. The Distribution of Phosphorus in Cow's milk In order to extract the phosphatides, it is necessary first to determine the distribution of phosphorus in cow's milk. Liquid milk can not be extracted with ether or with other organic solvents owing to its large content of water. The substance applied in this experiment is the milk powder, dried by the Buflovak drum drier below 70•Ž. Its content of moisture is 4.498%, deter mined by the method of drying in the air bath of 105•Ž. The content of total phosphorus was determined(5) in the ash of sample which was burned with a little of fusing mixture. In 100g. of milk powder In 100g. of dry matter Total phosphorus 0.745g. 0.781g. The amounts of phosphorus, which are soluble in the various organic sol vents, were determined by extracting with three solvents in the following orders. (A) Ether-Acetone-Alcohol (B) Acetone-Ether-Alcohol (C) Alcohol-Ether-Acetone Experiment (A) 1) At first a pair of each 10g. of sample was extracted with ether by the Soxhlet's apparatus for a week. After the extraction had been comp leted, the extract was freed from ether and weighed. In 100g.of milkpowder In 100g.of dry matter Total ether matters soluble 3.609g. 3.779g. 2) The above extract was dissolved in a very little of ether. -

United States Patent Office Patented Apr
3,030,421 United States Patent Office Patented Apr. 17, 1962 2 3,030,421 perature and under a reduced pressure. In the case of PROCESS FOR PREPARNG TRHYDROXY most of the catalysts the reaction products, which are free METHYL-PHOSPHINE s from solvents, solidify already when being cooled to room Martin Reuter and Ludwig Ortane, Frankfurt an Main, temperature. If, in the case of some catalysts, they Germany, assignors to Farbwerke Hoechst Aktienger 5 solidify only at a lower temperature and still contain to sellschaft vormas Meister Lucius & Briining, Frank a greater extent oily by-products and/or phosphonium furt am Main, Germany, a corporation of Gerinally hydroxide, they can be separated from the latter by filter No Drawing. Fied Jaa. 14, 1958, Ser. No. 708,764 ing or pressing. - - - Claims priority, application Germany Jan. 23, 1957 It is to be assumed that the crystalline main product 6 Canas. (C. 260-606.5) O of the present invention constitutes the hitherto unknown We have found that a new and valuable phosphorus trihydroxymethyl-phosphine. Main and by-products are compound carrying hydroxymethyl groups at the phos easily soluble in water and methanol and sparingly solu phorus atom can be prepared by reacting 1 mol of formal ble in fat dissolvers. dehyde with 4 mol of phosphine, preferably in the pres The reaction products of the invention can be used as ence of water, and in the presence of Small quantities of 5 insecticides, additives for lubricants, flame-proofing agents finely distributed metals that do not belong to the alkali for wood and textiles and as intermediates for these sub metals or alkaline earth metals and/or their compounds Staces. -

Thermodynamic Properties of Mixtures of Aqueous Solutions of Hydrochloric Acid and Cadmium Chloride, Copper Chloride, Manganese Chloride, and Zinc Chloride
Western Michigan University ScholarWorks at WMU Master's Theses Graduate College 12-1993 Thermodynamic Properties of Mixtures of Aqueous Solutions of Hydrochloric Acid and Cadmium Chloride, Copper Chloride, Manganese Chloride, and Zinc Chloride Samia A. Kosa Follow this and additional works at: https://scholarworks.wmich.edu/masters_theses Part of the Chemistry Commons Recommended Citation Kosa, Samia A., "Thermodynamic Properties of Mixtures of Aqueous Solutions of Hydrochloric Acid and Cadmium Chloride, Copper Chloride, Manganese Chloride, and Zinc Chloride" (1993). Master's Theses. 4363. https://scholarworks.wmich.edu/masters_theses/4363 This Masters Thesis-Open Access is brought to you for free and open access by the Graduate College at ScholarWorks at WMU. It has been accepted for inclusion in Master's Theses by an authorized administrator of ScholarWorks at WMU. For more information, please contact [email protected]. THERMODYNAMIC PROPERTIES OF MIXTURES OF AQUEOUS SOLUTIONS OF HYDROCHLORIC ACID AND CADMIUM CHLORIDE, COPPER CHLORIDE, MANGANESE CHLORIDE, AND ZINC CHLORIDE by Samia A. Kosa A Thesis Submitted to the Faculty of The Graduate College in partial fulfillment of the requirements for the Degree of Master of Arts Department of Chemistry Western Michigan University Kalamazoo, Michigan December 1993 ACKNOWLEDGEMENTS I wish to express my deep gratitude and sincere appreciation to my committee advisor, Dr. Donald Schriber, for his continual guidance, direction, and assistance in this research. I would also like to acknowledge the help of the other committee members, Dr. Thomass Houser and Dr. Ralph Steinhaus, towards this work. My family continuously provided me support and encouragement and to them I owe more than I can say. -

Download Author Version (PDF)
Journal of Materials Chemistry A Accepted Manuscript This is an Accepted Manuscript, which has been through the Royal Society of Chemistry peer review process and has been accepted for publication. Accepted Manuscripts are published online shortly after acceptance, before technical editing, formatting and proof reading. Using this free service, authors can make their results available to the community, in citable form, before we publish the edited article. We will replace this Accepted Manuscript with the edited and formatted Advance Article as soon as it is available. You can find more information about Accepted Manuscripts in the Information for Authors. Please note that technical editing may introduce minor changes to the text and/or graphics, which may alter content. The journal’s standard Terms & Conditions and the Ethical guidelines still apply. In no event shall the Royal Society of Chemistry be held responsible for any errors or omissions in this Accepted Manuscript or any consequences arising from the use of any information it contains. www.rsc.org/materialsA Page 1 of 9 Journal of Materials Chemistry A ARTICLE JMCA Safer Salts for CdTe Nanocrystal Solution Processed Solar Cells: The Dual Roles of Ligand Exchange and Grain Growth Received 00th January 20xx, a b c d e Accepted 00th January 20xx Troy K. Townsend, † William B. Heuer, Edward E. Foos, Eric Kowalski, Woojun Yoon and Joseph G. Tischler e DOI: 10.1039/x0xx00000x Inorganic CdSe/CdTe nanocrystals for solid-state photovoltaic devices are typically sintered into a bulk-like material after www.rsc.org/ annealing in the presence of solid cadmium chloride. -

Pearson-CV-15.Pdf
A. J. Pearson/C.V. January 2016 Curriculum Vitae Anthony J. Pearson Academic Rank: Rudolph & Susan Rense Professor of Chemistry Birthplace: Kingswinford, England. Citizenship: British; U.S. Naturalized Citizen. Education: University of Leeds, England, B.Sc. Hons. Class 1, 1971, Chemistry. Aston University, England, Ph.D., 1974, Organic Chemistry. Awards & Honors: Akroyd Scholarship (1969), Whytlaw-Gray Prize for Chemistry (1971), and Dawson Prize for Physical Chemistry (1971), University of Leeds. Sir Gilbert Morgan Medal (1973), Society for Chemical Industry, U.K. Science and Engineering Research Council (U.K.) Advanced Fellowship (1977-82). Sigma Xi Research Award (1984), Case Western Reserve University. John S. Diekhoff Award for Distinguished Graduate Teaching (1994), Case Western Reserve University. Visiting Scientist, Chemistry Research Promotion Center, Taipei, Taiwan, R.O.C., May 1990. Visiting Professor, University of Auckland, New Zealand, July/August, 1995. Chairman-Elect, American Chemical Society, Cleveland Section, 1999. Chairman, 2000. Finalist (one of three) for Northern Ohio Live 2001 Award of Achievement in Architecture, with R. Bostwick, N. Distad, K. Kutina, and N. Rushforth, for design of Agnar Pytte Science Center at CWRU. Case Alumni Association, Recognition of Meritorious Service, 2003. Experience: 1963-66 Chemist. Industrial research and analytical laboratories. Albright & Wilson; British Steel; West Midlands Gas Board. Semi-professional musician. 1974-77 Postdoctoral Research Fellow, with Arthur J. Birch. Research School of Chemistry, Australian National University, Canberra, Australia. 1977-82 SERC Advanced Fellow. Cambridge University Chemical Laboratory. 1978-81 Pauline Merz Official Fellow and Lecturer in Chemistry. Girton College, Cambridge University. 1979-81 Tutor. Girton College, Cambridge University. 1982-84 Associate Professor of Chemistry. -
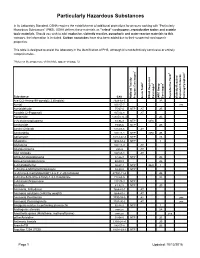
Particularly Hazardous Substances
Particularly Hazardous Substances In its Laboratory Standard, OSHA requires the establishment of additional protections for persons working with "Particularly Hazardous Substances" (PHS). OSHA defines these materials as "select" carcinogens, reproductive toxins and acutely toxic materials. Should you wish to add: explosive, violently reactive, pyrophoric and water-reactve materials to this category, the information is included. Carbon nanotubes have also been added due to their suspected carcinogenic properties. This table is designed to assist the laboratory in the identification of PHS, although it is not definitively conclusive or entirely comprehensive. *Notes on the proper use of this table appear on page 12. 1 6 5 2 3 4 Substance CAS National Toxicity National Program Carcinogen Toxin Acute Regulated OSHA Carcinogen Group IARC Carcinogen Toxin Reproductive Violently Reactive/ Explosive/Peroxide Forming/Pyrophoric A-a-C(2-Amino-9H-pyrido[2,3,b]indole) 2648-68-5 2B Acetal 105-57-7 yes Acetaldehyde 75-07-0 NTP AT 2B Acrolein (2-Propenal) 107-02-8 AT Acetamide 126850-14-4 2B 2-Acetylaminofluorene 53-96-3 NTP ORC Acrylamide 79-06-6 NTP 2B Acrylyl Chloride 814-68-6 AT Acrylonitrile 107-13-1 NTP ORC 2B Adriamycin 23214-92-8 NTP 2A Aflatoxins 1402-68-2 NTP 1 Allylamine 107-11-9 AT Alkylaluminums varies AT Allyl Chloride 107-05-1 AT ortho-Aminoazotoluene 97-56-3 NTP 2B para-aminoazobenzene 60-09-3 2B 4-Aminobiphenyl 92-67-1 NTP ORC 1 1-Amino-2-Methylanthraquinone 82-28-0 NTP (2-Amino-6-methyldipyrido[1,2-a:3’,2’-d]imidazole) 67730-11-4 2B -

The Effect of Sargassum Angustifolium Ethanol Extract on Cadmium Chloride-Induced Hypertension in Rat
Research Journal of Pharmacognosy (RJP) 8(1), 2021: 81-89 Received: 31 Oct 2020 Accepted: 17 Dec 2020 Published online: 19 Dec 2020 DOI: 10.22127/RJP.2020.255203.1637 Original article The Effect of Sargassum angustifolium Ethanol Extract on Cadmium Chloride-Induced Hypertension in Rat Leila Safaeian1* , Afsaneh Yegdaneh2, Masoud Mobasherian1 1Department of Pharmacology and Toxicology, Isfahan Pharmaceutical Sciences Research Center, School of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran. 2Department of Pharmacognosy, School of Pharmacy and Pharmaceutical Sciences, Isfahan University of Medical Sciences, Isfahan, Iran. Abstract Background and objectives: Sargassum angustifolium is a brown alga in southwestern coastline of Persian Gulf. Regarding the presence of various bioactive compounds and evidence of antihypertensive effects in other species of Sargassum, we evaluated the effect of S. angustifolium ethanol extract in CdCl2-induced hypertension in Wistar rats. Methods: Alga extract was prepared by maceration method using 70% ethanol and assessed for total phenolics and salt content. CdCl2 (1.5 mg/kg/day) was administered intraperitoneally to the rats for two weeks. Treatment groups received S. angustifolium extract (20, 40 and 80 mg/kg) or nifedipine (10 mg/kg) orally and simultaneously were given CdCl2 for two weeks. Systolic blood pressure (SBP) and heart rate were measured using tail-cuff method. Total antioxidant capacity, urea, creatinine, electrolytes including sodium, potassium, calcium and chloride were estimated in blood samples. The weight and histopathology of kidney tissues were also evaluated. Results: The content of total phenolic as gallic acid equivalent and the salt as NaCl was 67.42 ± 9.5 mg/g and 6.9 g/100 g in dried ethanol extract, respectively. -

Safety Data Sheet
SAFETY DATA SHEET Creation Date 27-Aug-2013 Revision Date 14-Feb-2020 Revision Number 2 1. Identification Product Name Cadmium bromide, ultra dry Cat No. : 47105 CAS-No 7789-72-6 Synonyms No information available Recommended Use Laboratory chemicals. Uses advised against Food, drug, pesticide or biocidal product use. Details of the supplier of the safety data sheet Company Alfa Aesar Thermo Fisher Scientific Chemicals, Inc. 30 Bond Street Ward Hill, MA 01835-8099 Tel: 800-343-0660 Fax: 800-322-4757 Email: [email protected] www.alfa.com Emergency Telephone Number During normal business hours (Monday-Friday, 8am-7pm EST), call (800) 343-0660. After normal business hours, call Carechem 24 at (866) 928-0789. 2. Hazard(s) identification Classification This chemical is considered hazardous by the 2012 OSHA Hazard Communication Standard (29 CFR 1910.1200) Acute oral toxicity Category 4 Acute dermal toxicity Category 4 Acute Inhalation Toxicity - Dusts and Mists Category 4 Carcinogenicity Category 1A Label Elements Signal Word Danger Hazard Statements May cause cancer Harmful if swallowed, in contact with skin or if inhaled ______________________________________________________________________________________________ Page 1 / 7 Cadmium bromide, ultra dry Revision Date 14-Feb-2020 ______________________________________________________________________________________________ Precautionary Statements Prevention Obtain special instructions before use Do not handle until all safety precautions have been read and understood Use personal protective equipment -

Pp-03-25-New Dots.Qxd 10/23/02 2:16 PM Page 136
pp-03-25-new dots.qxd 10/23/02 2:16 PM Page 136 136 BROMIC ACID / BROMINE BROMIC ACID [7789–31–3] Formula: HBrO3; MW 128.91 Uses Bromic acid is used as an oxidizing agent; and also as intermediate in the preparation of dyes and pharmaceuticals . Physical Properties Unstable compound; stable only in dilute aqueous solutions; solution turns yellow on standing; decomposes when heated to 100°C. Preparation Bromic acid is prepared by adding sulfuric acid to barium bromate. Ba(BrO3)2 + H2SO4 → 2HBrO3 + BaSO4 The product is distilled and absorbed in water. A 50% solution may be obtained by slow evaporation of the dilute solution in vacuum at –12°C. Toxicity Contact with skin and eyes can cause severe irritation. BROMINE [7726–95–6] Symbol Br; atomic number 35; atomic weight 79.904; a halogen group ele- ment; electron affinity 3.36359 eV; electronegativity 2.8; electron configura- tion [Ar] 3d104s24p5; most stable valence states –1 and +5, less stable valence states +1 and +3; a diatomic molecule (Br2) in liquid and vapor states over a wide range of temperature; two stable isotopes, Br–79 (50.57%) and Br–81 (49.43%). Occurrence and Uses Bromine occurs in nature as bromide in many natural brine wells and salt deposits. It also is found in seawater at a concentration of 85 mg/L. The ele- ment was discovered by A. J. Balard and C. Lowig, independently in 1826. Bromine is used in bleaching fibers and as a disinfectant for water purifica- tion. Other applications are in organic synthesis as an oxidizing or brominat- ing agent; in the manufacture of ethylene dibromide, methyl bromide and other bromo compounds for dyes and pharmaceutical uses; as a fire retardant for plastics; and in chemical analysis. -

Table of Contents
UNIVERSITY OF MISSOURI-KANSAS CITY CHEMICAL MANAGEMENT PLAN Revised May 2016 UMKC CHEMICAL MANAGEMENT PLAN This document constitutes the Chemical Management Plan (CMP) for the University of Missouri-Kansas City (UMKC). It was developed by the Environmental Health and Safety Department (EHS), to ensure the safe and proper use of hazardous and non- hazardous chemicals and to comply with applicable governmental regulations addressing the disposal of these chemicals. In addition, it was developed to foster waste minimization, and to provide the faculty and the staff with a management program to reduce the potential for accidents involving hazardous chemicals and/or wastes. Elements of the CMP include: a. a procedure for identifying potential or actual hazardous chemicals or wastes b. a procedure for periodic reexamination of those hazardous chemicals or wastes identified by the procedure in (a.) above as well as a systematic method for identification and evaluation of any new potential or actual hazardous chemicals or wastes c. procedures for labeling, and inventorying hazardous chemicals or wastes d. a procedure for identification and training of personnel directly responsible for ensuring that (a.), (b.), and (c.) are implemented e. a procedure for monitoring, recording, and reporting compliance with the CMP f. a procedure by which information generated by the CMP is provided to the persons performing waste analyses Each element is addressed as part of the complete CMP in the following paragraphs. 4 Table of Contents 1 Definitions 7 2 Identification -

Attachment 3-1 Guidance for Developing Ecological Soil
Attachment 3-1 Guidance for Developing Ecological Soil Screening Levels (Eco-SSLs) Eco-SSL Standard Operating Procedure (SOP # 1): Plant and Soil Invertebrate Literature Search and Acquisition OSWER Directive 92857-55 November 2003 This page intentionally left blank OVERVIEW Currently, there is a lack of clear guidance in setting terrestrial effect thresholds when conducting risk assessments. Without an EPA-approved, peer-reviewed, ecologically-based terrestrial effect database, the process to develop thresholds is problematic both to EPA, other federal agencies, states, and concerned private parties. Identification of published toxicity studies on invertebrates, microbial processes and plants is a key step in the derivation of benchmarks. The purpose of the Task Group 4, Standard Operating Procedure Number 1: Literature Search and Acquisition (referred to as TG4-SOP#1) is to document procedures used to identify and acquire potentially relevant toxicology literature for use in setting ecological soil screening levels. The literature search strategy is designed to locate worldwide terrestrial toxicity literature that includes the effects of chemicals of concern on terrestrial soil-dwelling invertebrates and plants. The literature acquisition process is designed to ensure timely acquisition of relevant publications. LITERATURE IDENTIFICATION Potentially relevant literature for developing ecological soil screening levels (Eco-SSLs) is identified by examining hard copies of relevant journals, bibliographies and guidance publications and through the use of a comprehensive computerized literature search strategy. These procedures are designed to locate worldwide terrestrial toxicology literature that includes the effects of specific toxic substances with an emphasis on exposure via soil. Paper-based Literature Identification The paper-based literature identification process includes the scanning of relevant review article bibliographies and key journals held in the U.S. -

The Direct Electrochemical Synthesis of Selected Group-Iib Organometallic Compounds
University of Windsor Scholarship at UWindsor Electronic Theses and Dissertations Theses, Dissertations, and Major Papers 1-1-1979 THE DIRECT ELECTROCHEMICAL SYNTHESIS OF SELECTED GROUP-IIB ORGANOMETALLIC COMPOUNDS. AKHTAR OSMAN University of Windsor Follow this and additional works at: https://scholar.uwindsor.ca/etd Recommended Citation OSMAN, AKHTAR, "THE DIRECT ELECTROCHEMICAL SYNTHESIS OF SELECTED GROUP-IIB ORGANOMETALLIC COMPOUNDS." (1979). Electronic Theses and Dissertations. 7187. https://scholar.uwindsor.ca/etd/7187 This online database contains the full-text of PhD dissertations and Masters’ theses of University of Windsor students from 1954 forward. These documents are made available for personal study and research purposes only, in accordance with the Canadian Copyright Act and the Creative Commons license—CC BY-NC-ND (Attribution, Non-Commercial, No Derivative Works). Under this license, works must always be attributed to the copyright holder (original author), cannot be used for any commercial purposes, and may not be altered. Any other use would require the permission of the copyright holder. Students may inquire about withdrawing their dissertation and/or thesis from this database. For additional inquiries, please contact the repository administrator via email ([email protected]) or by telephone at 519-253-3000ext. 3208. 492-33 National Library Bibliothfeque nationale CANADIAN THESES THESES CANADtENNES f l * .of Canada ~ du Canada ON MICROFICHE SUB UJCBOFICHE NAME OF AUTHOR/NOM O f L’AUTEUR. A khtar Osman TITLE OF THEStS/7777?£ O f LA THESE- The direct electrochemical synthesis of selected grouo IIB organonetallic compounds. UNIVERSITY/LW/ VERS/ TE. U niversity of ~Windsor. Windsor ..Ontario DEGREE FOR WHICH THESIS WAS PRESENTED/.