Bcats 2000 Symposium Proceedings
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Flexible Fluidic Actuators for Soft Robotic Applications
University of Wollongong Research Online University of Wollongong Thesis Collection 2017+ University of Wollongong Thesis Collections 2019 Flexible Fluidic Actuators for Soft Robotic Applications Weiping Hu Follow this and additional works at: https://ro.uow.edu.au/theses1 University of Wollongong Copyright Warning You may print or download ONE copy of this document for the purpose of your own research or study. The University does not authorise you to copy, communicate or otherwise make available electronically to any other person any copyright material contained on this site. You are reminded of the following: This work is copyright. Apart from any use permitted under the Copyright Act 1968, no part of this work may be reproduced by any process, nor may any other exclusive right be exercised, without the permission of the author. Copyright owners are entitled to take legal action against persons who infringe their copyright. A reproduction of material that is protected by copyright may be a copyright infringement. A court may impose penalties and award damages in relation to offences and infringements relating to copyright material. Higher penalties may apply, and higher damages may be awarded, for offences and infringements involving the conversion of material into digital or electronic form. Unless otherwise indicated, the views expressed in this thesis are those of the author and do not necessarily represent the views of the University of Wollongong. Research Online is the open access institutional repository for the University of -

Environmentally Controlled Curvature of Single Collagen Proteins
Environmentally controlled curvature of single collagen proteins Naghmeh Rezaei, Aaron Lyons and Nancy R. Forde* Department of Physics Simon Fraser University 8888 University Drive Burnaby, BC V5A 1S6 CANADA *Corresponding author. 778-782-3161. [email protected]. ORCID: 0000-0002-5479-7073 - 1 - ABSTRACT The predominant structural protein in vertebrates is collagen, which plays a key role in extracellular matrix and connective tissue mechanics. Despite its prevalence and physical importance in biology, the mechanical properties of molecular collagen are far from established. The flexibility of its triple helix is unresolved, with descriptions from different experimental techniques ranging from flexible to semirigid. Furthermore, it is unknown how collagen type (homo- vs. heterotrimeric) and source (tissue-derived vs. recombinant) influence flexibility. Using SmarTrace, a chain tracing algorithm we devised, we performed statistical analysis of collagen conformations collected with atomic force microscopy (AFM) to Our results show that types I, II and III collagens the key fibrillar varieties exhibit molecular flexibilities that are very similar. However, collagen conformations are strongly modulated by salt, transitioning from compact to extended as KCl concentration increases, in both neutral and acidic pH. While analysis with a standard worm-like chain model suggests that the persistence length of collagen can attain almost any value within the literature range, closer inspection reveals that this modulation to changes in flexibility, but rather arises from the induction of curvature (either intrinsic or induced by interactions with the mica surface). By modifying standard polymer theory to include innate curvature, we show that collagen behaves as an equilibrated curved worm-like chain (cWLC) in two dimensions. -

Peptoid Residues Make Diverse, Hyperstable Collagen Triple Helices
Peptoid Residues Make Diverse, Hyperstable Collagen Triple Helices Julian L. Kessler1, Grace Kang1, Zhao Qin2, Helen Kang1, Frank G. Whitby3, Thomas E. Cheatham III4, Christopher P. Hill3, Yang Li1,*, and S. Michael Yu1,5 1Department of Biomedical Engineering, University of Utah, Salt Lake City, Utah 84112, USA 2Department of Civil & Environmental Engineering, Collagen of Engineering & Computer Science, Syracuse University, Syracuse, New York 13244, USA 3Department of Biochemistry, University of Utah School of Medicine, Salt Lake City, UT 84112, USA 4Department of Medicinal Chemistry, College of Pharmacy, L. S. Skaggs Pharmacy Research Institute, University of Utah, Salt Lake City, Utah 84112, USA 5Department of Pharmaceutics and Pharmaceutical Chemistry, University of Utah, Salt Lake City, Utah 84112, USA *Corresponding Author: Yang Li ([email protected]) Abstract The triple-helical structure of collagen, responsible for collagen’s remarkable biological and mechanical properties, has inspired both basic and applied research in synthetic peptide mimetics for decades. Since non-proline amino acids weaken the triple helix, the cyclic structure of proline has been considered necessary, and functional collagen mimetic peptides (CMPs) with diverse sidechains have been difficult to produce. Here we show that N-substituted glycines (N-glys), also known as peptoid residues, exhibit a general triple-helical propensity similar to or greater than proline, allowing synthesis of thermally stable triple-helical CMPs with unprecedented sidechain diversity. We found that the N-glys stabilize the triple helix by sterically promoting the preorganization of individual CMP chains into the polyproline-II helix conformation. Our findings were supported by the crystal structures of two atomic-resolution N-gly-containing CMPs, as well as experimental and computational studies spanning more than 30 N-gly-containing peptides. -
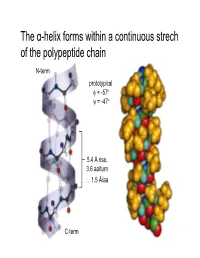
The Α-Helix Forms Within a Continuous Strech of the Polypeptide Chain
The α-helix forms within a continuous strech of the polypeptide chain N-term prototypical φ = -57 ° ψ = -47 ° 5.4 Å rise, 3.6 aa/turn ∴ 1.5 Å/aa C-term α-Helices have a dipole moment, due to unbonded and aligned N-H and C=O groups β-Sheets contain extended (β-strand) segments from separate regions of a protein prototypical φ = -139 °, ψ = +135 ° prototypical φ = -119 °, ψ = +113 ° (6.5Å repeat length in parallel sheet) Antiparallel β-sheets may be formed by closer regions of sequence than parallel Beta turn Figure 6-13 The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups Helix wheel The stability of helices and sheets depends on their sequence of amino acids • Intrinsic propensity of an amino acid to adopt a helical or extended (strand) conformation • Interactions between adjacent R-groups – Ionic attraction or repulsion – Steric hindrance of adjacent bulky groups • Occurrence of proline and glycine • Interactions between ends of helix and aa R-groups His Glu N-term C-term -

Y. Ma, X. Feng, J.A. Rogers, Y. Huang and Yihui
Lab on a Chip View Article Online CRITICAL REVIEW View Journal | View Issue Design and application of ‘J-shaped’ stress–strain Cite this: Lab Chip,2017,17,1689 behavior in stretchable electronics: a review Yinji Ma,ab Xue Feng,a John A. Rogers,c Yonggang Huangb and Yihui Zhang *a A variety of natural biological tissues (e.g., skin, ligaments, spider silk, blood vessel) exhibit ‘J-shaped’ stress–strain behavior, thereby combining soft, compliant mechanics and large levels of stretchability, with anatural‘strain-limiting’ mechanism to prevent damage from excessive strain. Synthetic materials with similar stress–strain behaviors have potential utility in many promising applications, such as tissue engineer- ing (to reproduce the nonlinear mechanical properties of real biological tissues) and biomedical devices (to enable natural, comfortable integration of stretchable electronics with biological tissues/organs). Recent advances in this field encompass developments of novel material/structure concepts, fabrication ap- proaches, and unique device applications. This review highlights five representative strategies, including de- signs that involve open network, wavy and wrinkled morphologies, helical layouts, kirigami and origami constructs, and textile formats. Discussions focus on the underlying ideas, the fabrication/assembly routes, Received 18th March 2017, and the microstructure–property relationships that are essential for optimization of the desired ‘J-shaped’ Accepted 21st April 2017 stress–strain responses. Demonstration applications provide examples of the use of these designs in de- formable electronics and biomedical devices that offer soft, compliant mechanics but with inherent robust- DOI: 10.1039/c7lc00289k ness against damage from excessive deformation. We conclude with some perspectives on challenges and rsc.li/loc opportunities for future research. -

Gifc-2018 Book of Abstracts
GIFC-2018 Giornate Italo-Francesi di Chimica Journées Franco-Italiennes de Chimie 16 – 18 April 2018 Grand Hotel Savoia Via Arsenale di Terra, 5 Genova BOOK OF ABSTRACTS ISBN: 978-88-94952-00-1 Gold Sponsors Silver Sponsors Patronages 2 3 Scientific Program 4 5 6 List of Posters (according to alphabetic order of presenting Authors) 1st session, Monday 16th April Num Cognome Nome Contatto Titolo Synthesis, purification and characterization of PO1 Aboudou Soioulata [email protected] antimicrobial peptides isolated from animal venoms [email protected] PO2 Ajmalghan Muthali Coverage dependent recombination mechanisms of hydrogen from niv-amu.fr tungsten surfaces via density functional theory Hybrid squaraine-silica nanoparticles as nir probes for biological PO3 Alberto Gabriele [email protected] applications: optimization of the photoemission performances Synthesis of doped metal oxide nanocrystals for solution-processed PO4 Alkarsifi Riva [email protected] interfacial layers in organic solar cells PO5 Anceschi Anastasia [email protected] Maltodextrins nanosponges as precursor for porous carbon materials [email protected] PO7 Arnodo Davide First racemic total synthesis of heliolactone .it [email protected]. PO8 Azzi Emanuele Synthesis of boronated analogue of curcumin as potential therapeutical it agents for alzheimer’s disease Study of Nanobiomaterials with Bio-based Antioxidants: Interaction of PO9 Barzan Giulia Sci piem Polyphenol Molecules with Hydroxyapatite and Silica Luigi Methotrexate loaded solid lipid nanoparticles: protein functionalization to PO10 Battaglia [email protected] Sebastiano improve brain biodistribution [email protected] PO11 Begni Federico On the adsorption of toluene on porous materials with different chemical m composition Synthesischaracterization of activated carbon from modified banana peels PO12 Ben Khalifa Eya [email protected] for hexavalent chromium adsorption [email protected] PO13 Benvenuti Martino The maturation of the co-dehydrogenase from thermococcus sp. -

Flexible Fluidic Actuators for Soft Robotic Applications
University of Wollongong Research Online University of Wollongong Thesis Collection 2017+ University of Wollongong Thesis Collections 2019 Flexible Fluidic Actuators for Soft Robotic Applications Weiping Hu University of Wollongong Follow this and additional works at: https://ro.uow.edu.au/theses1 University of Wollongong Copyright Warning You may print or download ONE copy of this document for the purpose of your own research or study. The University does not authorise you to copy, communicate or otherwise make available electronically to any other person any copyright material contained on this site. You are reminded of the following: This work is copyright. Apart from any use permitted under the Copyright Act 1968, no part of this work may be reproduced by any process, nor may any other exclusive right be exercised, without the permission of the author. Copyright owners are entitled to take legal action against persons who infringe their copyright. A reproduction of material that is protected by copyright may be a copyright infringement. A court may impose penalties and award damages in relation to offences and infringements relating to copyright material. Higher penalties may apply, and higher damages may be awarded, for offences and infringements involving the conversion of material into digital or electronic form. Unless otherwise indicated, the views expressed in this thesis are those of the author and do not necessarily represent the views of the University of Wollongong. Recommended Citation Hu, Weiping, Flexible Fluidic Actuators for Soft Robotic Applications, Doctor of Philosophy thesis, School of Mechanical, Materials, Mechatronic and Biomedical Engineering, University of Wollongong, 2019. https://ro.uow.edu.au/theses1/717 Research Online is the open access institutional repository for the University of Wollongong. -

Daftar Peneliti Yang Belum Mengunggah Laporan Anggaran Dan Laporan Akhir Tahun 2014
LAMPIRAN SURAT NO : 4033/E5.1/PE/2014 DAFTAR PENELITI YANG BELUM MENGUNGGAH LAPORAN ANGGARAN DAN LAPORAN AKHIR TAHUN 2014 NO NAMA NIDN PERGURUAN TINGGI JUDUL SKIM LAPORAN ANGGARAN LAPORAN AKHIR Akademi Analis Kesehatan ANALISIS KUALITAS DAN TINGKAT OKSIDASI MINYAK 1 LISA ANDINA 1123038303 Dosen Pemula belum diunggah belum diunggah Borneo Lestari Banjarbaru GORENG CURAH YANG DIGUNAKAN PADA WARUNG Akademi Analis Kesehatan PengembanganMAKAN SEAFOOD Produk KAKI LIMAHerbal MENGGUNAKAN Terstandar Sebagai 2 FAISAL 0710117901 Dosen Pemula belum diunggah belum diunggah Malang Obat Antimalaria dari Ekstrak daun Tanaman Bunga Akademi Analis Kesehatan PENGARUHmatahari (Helianthus AGONIS PEROXISOME annus) PROLIFERATOR- 3 NUGROHO TRISTYANTO 0713068101 Dosen Pemula belum diunggah sudah diunggah Malang ACTIVATED RECEPTOR γ (PPARγ) TERHADAP PRODUKSI Akademi Analis Kesehatan ISOLASIVASCULAR DAN ENDOTHELIAL ELUSIDASI STRUKTUR GROWTH SENYAWAFACTOR (VEGF) 4 ROIHATUL MUTIAH 0703028003 Pekerti belum diunggah sudah diunggah Malang ANTIKANKER DARI DAUN DAN BUNGA Calotrophis Akademi Analis Kesehatan Pengaruhgigantea SERTAKualitas PENENTUAN Tidur Dengan MEKANISME Jumlah Sel Darah 5 CISILLIA ADHIYANI 0623038004 Dosen Pemula belum diunggah belum diunggah Nasional Surakarta Pekerja Dengan Sistem Shift Akademi Bahasa Asing RA PENGEMBANGAN MODEL RELOKASI 6 AGUS DWIDOSO WARSONO 0626086502 Disertasi Doktor belum diunggah belum diunggah Kartini Surakarta PEDAGANG KAKI LIMA DALAM UPAYA REVITALISASI CRESCENTIANA EMY Akademi Farmasi Nasional PengaruhPEDAGANG Cara KAKI Pemanfaatan -

Triple‐Helix‐Stabilizing Effects in Collagen Model Peptides
Angewandte Research Articles Chemie International Edition:DOI:10.1002/anie.201914101 Protein Folding German Edition:DOI:10.1002/ange.201914101 Triple-Helix-Stabilizing Effects in Collagen Model Peptides Containing PPII-Helix-Preorganized Diproline Modules Andreas Maaßen, JanM.Gebauer,Elena Theres Abraham, Isabelle Grimm, Jçrg- Martin Neudçrfl, Ronald Kühne,Ines Neundorf,Ulrich Baumann,* and Hans- Günther Schmalz* Abstract: Collagen model peptides (CMPs) serve as tools for guarantees the structural integrity of vertebrates.[1–3] In understanding stability and function of the collagen triple helix addition, collagen interacts with numerous proteins such as and have apotential for biomedical applications.Inthe past, cell-surface receptors or matrix metalloproteinases and is interstrand cross-linking or conformational preconditioning of involved in processes such as cell adhesion, proliferation, and proline units through stereoelectronic effects have been utilized extracellular matrix regulation.[4,5] Thebiocompatibility and in the design of stabilized CMPs.Tofurther study the effects bioactivity of collagen together with advances in synthetic determining collagen triple helix stability we investigated substitutes[6,7] open the path for biomedical applications such aseries of CMPs containing synthetic diproline-mimicking as bone grafts,[8,9] wound dressings,[10,11] engineering of func- modules (ProMs), whichwere preorganized in aPPII-helix- tional tissues,[12,13] tendon repair,[14] or inhibition of disease- [4,15] type conformation by afunctionalizable -

This Deleuzian Century Faux Titre Etudes De Langue Et Littérature Françaises
This Deleuzian Century Faux Titre Etudes de langue et littérature françaises Series Editors Keith Busby M.J. Freeman† Sjef Houppermans et Paul Pelckmans VOLUME 400 The titles published in this series are listed at brill.com/faux This Deleuzian Century Art, Activism, Life Edited by Rosi Braidotti Rick Dolphijn LEIDEN | BOSTON Cover illustration: Denis Sinyakov, photographer, www.denissinyakov.com Library of Congress Control Number: 2015933470 issn 0167-9392 isbn 978-90-42-03916-2 (paperback) isbn 978-94-01-21198-7 (e-book) Copyright 2014 by Koninklijke Brill nv, Leiden, The Netherlands. Koninklijke Brill nv incorporates the imprints Brill, Brill Hes & De Graaf, Brill Nijhoff, Brill Rodopi and Hotei Publishing. All rights reserved. No part of this publication may be reproduced, translated, stored in a retrieval system, or transmitted in any form or by any means, electronic, mechanical, photocopying, recording or otherwise, without prior written permission from the publisher. Authorization to photocopy items for internal or personal use is granted by Koninklijke Brill nv provided that the appropriate fees are paid directly to The Copyright Clearance Center, 222 Rosewood Drive, Suite 910, Danvers, ma 01923, usa. Fees are subject to change. This book is printed on acid-free paper. Acknowledgments As the editors of This Deleuzian Century, we wish to specially thank the following persons for hosting the National Symposium on Deleuze Scholarship: Andrej Radman, Sjoerd van Tuinen, Henk Oosterling, Frans- Willem Korsten, Arjen Kleinherenbrink and Anneke Smelik. Our gratitude to Denis Sinyakov for donating the striking photograph on the book cover. Many thanks to Christa Stevens from Rodopi for her editorial assistance in the preparation of this volume; to Sophie Chapple for helping with the copy editing process, and especially to Goda Klumbyte and Toa Maes for general assistance. -

Mechanics and Structural Stability of the Collagen Triple Helix
Mechanics and Structural Stability of the Collagen Triple Helix Michael W.H. Kirkness,1 Kathrin Lehmann2 and Nancy R. Forde1,2 1Department of Molecular Biology and Biochemistry and 2Department of Physics, Simon Fraser University. Burnaby, BC, V5A 1S6 Canada [email protected] Abstract The primary building block of the body is collagen, which is found in the extracellular matrix and in many stress-bearing tissues such as tendon and cartilage. It provides elasticity and support to cells and tissues while influencing biological pathways including cell signaling, motility and differentiation. Collagen’s unique triple helical structure is thought to impart mechanical stability. However, detailed experimental studies on its molecular mechanics have been only recently emerging. Here, we review the treatment of the triple helix as a homogeneous flexible rod, including bend (standard worm-like chain model), twist, and stretch deformations, and the assumption of backbone linearity. Additionally, we discuss protein- specific properties of the triple helix including sequence dependence, and relate single-molecule mechanics to collagen’s physiological context. Introduction Collagen is the most abundant protein in the human body, accounting for more than 30% of the total protein.1,2 Collagens are defined by their unique right-handed triple helical structure, comprised of three left-handed polyproline-like helices, each with a (Gly-X-Y) repeating sequence where X and Y are often proline and hydroxyproline.2 In the body, collagen forms ordered, hierarchical -

Abstract Book | Seattle, WA | 1-4 November 2018
Abstract Book | Seattle, WA | 1-4 November 2018 Table of Contents_Toc528229514 “A Totally Unqualified Woman”: Gender and the Policing of Science in the IGY Expedition to South Georgia ...... 1 “Bringing the Bluegrass West: Scientific Agriculture and the California Thoroughbred Industry” ............................ 1 “China as a Field”: Carl Bishop, Ji Li, and the “First Archaeological Expedition in China led by the Chinese Themselves“ ............................................................................................................................................................. 1 “Collecting Evolution in the Galapagos and Rebuilding the California Academy of Sciences” .................................. 2 “Eating Electricity and Delivering India”: Cultural Resistance and Electricity in Late-nineteenth Century Bengali Drama ...................................................................................................................................................................... 2 “Esperienza,” Teacher of All Things: The Musical Art-Science of Vincenzo Galilei .................................................. 3 “Fossil Tug of War: Evolution and Controversy at Liang Bua” ................................................................................ 3 “How Do I Know... Prayers Don’t Do More Good than... Pills”: Don Pedrito Jaramillo, Curanderismo, and the Rise of Professional Medicine in the Texas-Mexico Borderlands over the Turn-of-the-Century ............................... 3 “Humanistic” Science, “Scientific” Humanities: