Myelin Interactions with Complement, Macrophages, and Microglia in Spinal Cord Injury Timothy J
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Myelin Biogenesis Is Associated with Pathological Ultrastructure That Is
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Myelin biogenesis is associated with pathological ultrastructure that 2 is resolved by microglia during development 3 4 5 Minou Djannatian1,2*, Ulrich Weikert3, Shima Safaiyan1,2, Christoph Wrede4, Cassandra 6 Deichsel1,2, Georg Kislinger1,2, Torben Ruhwedel3, Douglas S. Campbell5, Tjakko van Ham6, 7 Bettina Schmid2, Jan Hegermann4, Wiebke Möbius3, Martina Schifferer2,7, Mikael Simons1,2,7* 8 9 1Institute of Neuronal Cell Biology, Technical University Munich, 80802 Munich, Germany 10 2German Center for Neurodegenerative Diseases (DZNE), 81377 Munich, Germany 11 3Max-Planck Institute of Experimental Medicine, 37075 Göttingen, Germany 12 4Institute of Functional and Applied Anatomy, Research Core Unit Electron Microscopy, 13 Hannover Medical School, 30625, Hannover, Germany 14 5Department of Neuronal Remodeling, Graduate School of Pharmaceutical Sciences, Kyoto 15 University, Sakyo-ku, Kyoto 606-8501, Japan. 16 6Department of Clinical Genetics, Erasmus MC, University Medical Center Rotterdam, 3015 CN, 17 the Netherlands 18 7Munich Cluster of Systems Neurology (SyNergy), 81377 Munich, Germany 19 20 *Correspondence: [email protected] or [email protected] 21 Keywords 22 Myelination, degeneration, phagocytosis, microglia, oligodendrocytes, phosphatidylserine 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
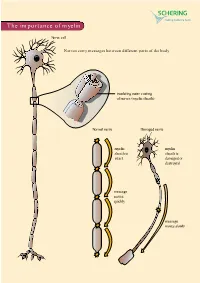
The Importance of Myelin
The importance of myelin Nerve cell Nerves carry messages between different parts of the body insulating outer coating of nerves (myelin sheath) Normal nerve Damaged nerve myelin myelin sheath is sheath is intact damaged or destroyed message moves quickly message moves slowly Nerve cells transmit impulses Nerve cells have a long, thin, flexible fibre that transmits impulses. These impulses are electrical signals that travel along the length of the nerve. Nerve fibres are long to enable impulses to travel between distant parts of the body, such as the spinal cord and leg muscles. Myelin speeds up impulses Most nerve fibres are surrounded by an insulating, fatty sheath called myelin, which acts to speed up impulses. The myelin sheath contains periodic breaks called nodes of Ranvier. By jumping from node to node, the impulse can travel much more quickly than if it had to travel along the entire length of the nerve fibre. Myelinated nerves can transmit a signal at speeds as high as 100 metres per second – as fast as a Formula One racing car. normal damaged nerve nerve Loss of myelin leads to a variety of symptoms If the myelin sheath surrounding nerve fibres is damaged or destroyed, transmission of nerve impulses is slowed or blocked. The impulse now has to flow continuously along the whole nerve fibre – a process that is much slower than jumping from node to node. Loss of myelin can also lead to ‘short-circuiting’ of nerve impulses. An area where myelin has been destroyed is called a lesion or plaque. This slowing and ‘short-circuiting’ of nerve impulses by lesions leads to a variety of symptoms related to nervous system activity. -

Regulation of Myelin Structure and Conduction Velocity by Perinodal Astrocytes
Correction NEUROSCIENCE Correction for “Regulation of myelin structure and conduc- tion velocity by perinodal astrocytes,” by Dipankar J. Dutta, Dong Ho Woo, Philip R. Lee, Sinisa Pajevic, Olena Bukalo, William C. Huffman, Hiroaki Wake, Peter J. Basser, Shahriar SheikhBahaei, Vanja Lazarevic, Jeffrey C. Smith, and R. Douglas Fields, which was first published October 29, 2018; 10.1073/ pnas.1811013115 (Proc. Natl. Acad. Sci. U.S.A. 115,11832–11837). The authors note that the following statement should be added to the Acknowledgments: “We acknowledge Dr. Hae Ung Lee for preliminary experiments that informed the ultimate experimental approach.” Published under the PNAS license. Published online June 10, 2019. www.pnas.org/cgi/doi/10.1073/pnas.1908361116 12574 | PNAS | June 18, 2019 | vol. 116 | no. 25 www.pnas.org Downloaded by guest on October 2, 2021 Regulation of myelin structure and conduction velocity by perinodal astrocytes Dipankar J. Duttaa,b, Dong Ho Wooa, Philip R. Leea, Sinisa Pajevicc, Olena Bukaloa, William C. Huffmana, Hiroaki Wakea, Peter J. Basserd, Shahriar SheikhBahaeie, Vanja Lazarevicf, Jeffrey C. Smithe, and R. Douglas Fieldsa,1 aSection on Nervous System Development and Plasticity, The Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892; bThe Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., Bethesda, MD 20817; cMathematical and Statistical Computing Laboratory, Office of Intramural Research, Center for Information -

View Software
TLR4-activated microglia have divergent effects on oligodendrocyte lineage cells DISSERTATION Presented in partial fulfillment of the requirements for the Degree Doctor of Philosophy in the Graduate School of The Ohio State University Evan Zachary Goldstein, BA Neuroscience Graduate Program The Ohio State University 2016 Dissertation Committee: Dr. Dana McTigue, Advisor Dr. Phillip Popovich Dr. Jonathan Godbout Dr. Courtney DeVries Copyright by Evan Zachary Goldstein 2016 i Abstract Myelin accelerates action potential conduction velocity and provides essential metabolic support for axons. Unfortunately, myelin and myelinating cells are often vulnerable to injury or disease, resulting in myelin damage, which in turn can lead to axon dysfunction, overt pathology and neurological impairment. Inflammation is a common component of CNS trauma and disease, and therefore an active inflammatory response is often considered deleterious to myelin health. While inflammation can certainly damage myelin, inflammatory processes also benefit oligodendrocyte (OL) lineage progression and myelin repair. Consistent with the divergent nature of inflammation, intraspinal toll-like receptor 4 (TLR4) activation, an innate immune pathway, kills OL lineage cells, but also initiates oligodendrogenesis. Soluble factors produced by TLR4-activated microglia can reproduce these effects in vitro, however the exact factors are unknown. To determine what microglial factors might contribute to TLR4-induced OL loss and oligodendrogenesis, mRNA of factors known to affect OL lineage cells was quantified in TLR4-activated microglia and spinal cords (chapter 2). Results indicate that TLR4-activated microglia transcribe numerous factors that induce OL loss, OL progenitor cell (OPC) proliferation and OPC differentiation. However, some factors upregulated after intraspinal TLR4 activation were not ii upregulated by microglia, suggesting that other cell types contribute to transcriptional changes in vivo. -
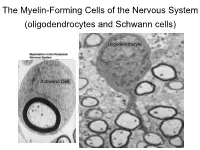
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Tecnicas Microscopicas
CAP 1: TÉCNICAS MICROSCÓPICAS TÉCNICAS MICROSCÓPICAS 11 Lic. Carlos R. Neira Montoya Lic. Eduardo Sedano Gelvet Lic. María Elena Vilcarromero V. El estudio de los tejidos tal como los observamos hoy en día no sería posible sin la ayuda de la histotecnología; esta disciplina se encarga del estudio de los métodos técnicas y procedimientos que permiten la transformación de un órgano en una película lo suficientemente transparente y contrastada que nos permite su observación a través del microscopio (Fig. 1-1). Para que esto ocurra se tiene que seguir una serie de pasos. Cada uno de estos pasos permite la observación de las características morfológicas del tejido que nos indica la normalidad o la alteración patológica; sin embargo en estudios mucho más minuciosos, estos pasos se harán en función de las estructuras o sustancias que se deseen investigar en la muestra correspondiente. Figura 1-1. Un órgano es transformado en una película transparente. - Pág. 5 - CAP 1: TÉCNICAS MICROSCÓPICAS Los pasos de las técnicas microscópicas para obtener un preparado histológico permanente (láminas) son: 1. Toma de la muestra. 2. Fijación. 3. Inclusión. 4. Microtomía. 5. Coloración. 1. TOMA DE LA MUESTRA Es el momento que se selecciona el órgano o tejido a estudiar. De tres fuentes puede provenir el material humano: las necropsias, las biopsias y las piezas operadas. De éstas, sólo la primera puede darnos material normal; las dos últimas habitualmente proporcionarán tejidos para estudio histopatológico. - Necropsias: son las piezas que se obtienen de un cadáver. Para histología normal es necesario que se trate de un cadáver fresco y que no haya sido atacado por ninguna lesión, por lo menos el órgano que se quiere estudiar. -

Nomina Histologica Veterinaria, First Edition
NOMINA HISTOLOGICA VETERINARIA Submitted by the International Committee on Veterinary Histological Nomenclature (ICVHN) to the World Association of Veterinary Anatomists Published on the website of the World Association of Veterinary Anatomists www.wava-amav.org 2017 CONTENTS Introduction i Principles of term construction in N.H.V. iii Cytologia – Cytology 1 Textus epithelialis – Epithelial tissue 10 Textus connectivus – Connective tissue 13 Sanguis et Lympha – Blood and Lymph 17 Textus muscularis – Muscle tissue 19 Textus nervosus – Nerve tissue 20 Splanchnologia – Viscera 23 Systema digestorium – Digestive system 24 Systema respiratorium – Respiratory system 32 Systema urinarium – Urinary system 35 Organa genitalia masculina – Male genital system 38 Organa genitalia feminina – Female genital system 42 Systema endocrinum – Endocrine system 45 Systema cardiovasculare et lymphaticum [Angiologia] – Cardiovascular and lymphatic system 47 Systema nervosum – Nervous system 52 Receptores sensorii et Organa sensuum – Sensory receptors and Sense organs 58 Integumentum – Integument 64 INTRODUCTION The preparations leading to the publication of the present first edition of the Nomina Histologica Veterinaria has a long history spanning more than 50 years. Under the auspices of the World Association of Veterinary Anatomists (W.A.V.A.), the International Committee on Veterinary Anatomical Nomenclature (I.C.V.A.N.) appointed in Giessen, 1965, a Subcommittee on Histology and Embryology which started a working relation with the Subcommittee on Histology of the former International Anatomical Nomenclature Committee. In Mexico City, 1971, this Subcommittee presented a document entitled Nomina Histologica Veterinaria: A Working Draft as a basis for the continued work of the newly-appointed Subcommittee on Histological Nomenclature. This resulted in the editing of the Nomina Histologica Veterinaria: A Working Draft II (Toulouse, 1974), followed by preparations for publication of a Nomina Histologica Veterinaria. -

Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Disease
MOG ANTIBODY DISEASE Myelin Oligodendrocyte Glycoprotein (MOG) Antibody-Associated Encephalomyelitis WHAT IS MOG ANTIBODY-ASSOCIATED DEMYELINATION? Myelin oligodendrocyte glycoprotein (MOG) antibody-associated demyelination is an immune-mediated inflammatory process that affects the central nervous system (brain and spinal cord). MOG is a protein that exists on the outer surface of cells that create myelin (an insulating layer around nerve fibers). In a small number of patients, an initial episode of inflammation due to MOG antibodies may be the first manifestation of multiple sclerosis (MS). Most patients may only experience one episode of inflammation, but repeated episodes of central nervous system demyelination can occur in some cases. WHAT ARE THE SYMPTOMS? • Optic neuritis (inflammation of the optic nerve(s)) may be a symptom of MOG antibody- associated demyelination, which may result in painful loss of vision in one or both eyes. • Transverse myelitis (inflammation of the spinal cord) may cause a variety of symptoms that include: o Abnormal sensations. Numbness, tingling, coldness or burning in the arms and/or legs. Some are especially sensitive to the light touch of clothing or to extreme heat or cold. You may feel as if something is tightly wrapping the skin of your chest, abdomen or legs. o Weakness in your arms or legs. Some people notice that they're stumbling or dragging one foot, or heaviness in the legs. Others may develop severe weakness or even total paralysis. o Bladder and bowel problems. This may include needing to urinate more frequently, urinary incontinence, difficulty urinating and constipation. • Acute disseminated encephalomyelitis (ADEM) may cause loss of vision, weakness, numbness, and loss of balance, and altered mental status. -

Targeting Myelin Lipid Metabolism As a Potential Therapeutic Strategy in a Model of CMT1A Neuropathy
ARTICLE DOI: 10.1038/s41467-018-05420-0 OPEN Targeting myelin lipid metabolism as a potential therapeutic strategy in a model of CMT1A neuropathy R. Fledrich 1,2,3, T. Abdelaal 1,4,5, L. Rasch1,4, V. Bansal6, V. Schütza1,3, B. Brügger7, C. Lüchtenborg7, T. Prukop1,4,8, J. Stenzel1,4, R.U. Rahman6, D. Hermes 1,4, D. Ewers 1,4, W. Möbius 1,9, T. Ruhwedel1, I. Katona 10, J. Weis10, D. Klein11, R. Martini11, W. Brück12, W.C. Müller3, S. Bonn 6,13, I. Bechmann2, K.A. Nave1, R.M. Stassart 1,3,12 & M.W. Sereda1,4 1234567890():,; In patients with Charcot–Marie–Tooth disease 1A (CMT1A), peripheral nerves display aberrant myelination during postnatal development, followed by slowly progressive demye- lination and axonal loss during adult life. Here, we show that myelinating Schwann cells in a rat model of CMT1A exhibit a developmental defect that includes reduced transcription of genes required for myelin lipid biosynthesis. Consequently, lipid incorporation into myelin is reduced, leading to an overall distorted stoichiometry of myelin proteins and lipids with ultrastructural changes of the myelin sheath. Substitution of phosphatidylcholine and phosphatidylethanolamine in the diet is sufficient to overcome the myelination deficit of affected Schwann cells in vivo. This treatment rescues the number of myelinated axons in the peripheral nerves of the CMT rats and leads to a marked amelioration of neuropathic symptoms. We propose that lipid supplementation is an easily translatable potential therapeutic approach in CMT1A and possibly other dysmyelinating neuropathies. 1 Department of Neurogenetics, Max-Planck-Institute of Experimental Medicine, Göttingen 37075, Germany. -

Microglia Activation Triggers Astrocyte-Mediated Modulation of Excitatory Neurotransmission
Microglia activation triggers astrocyte-mediated PNAS PLUS modulation of excitatory neurotransmission Olivier Pascuala,b,c,1,2, Sarrah Ben Achoura,b,c,2, Philippe Rostainga,b,c, Antoine Trillera,b,c, and Alain Bessisa,b,c aInstitut de Biologie de l’Ecole Normale Supérieure, F-75005 Paris, France; bInstitut National de la Santé et de la Recherche Médicale U1024, F-75005 Paris, France; and cCentre National de la Recherche Scientifique, Unité Mixte de Recherche 8197, F-75005 Paris, France Edited* by Tullio Pozzan, University of Padova, Padua, Italy, and approved November 21, 2011 (received for review July 18, 2011) Fine control of neuronal activity is crucial to rapidly adjust to subtle tatively able to sense neuronal activity and/or communicate with changes of the environment. This fine tuning was thought to be astrocytes. In response to stimuli, microglia are activated, and they purely neuronal until the discovery that astrocytes are active players release neurotransmitters (19), which are small molecules such as of synaptic transmission. In the adult hippocampus, microglia are nitric oxide, trophic factors, or cytokines, all known to control the other major glial cell type. Microglia are highly dynamic and neuronal function and synaptic transmission (20, 21). In addition, closely associated with neurons and astrocytes. They react rapidly to changes in plasticity and neuronal activity have been shown to modifications of their environment and are able to release mole- modify the resident time of microglia processes at synapses (22). cules known to control neuronal function and synaptic transmission. Although long-term effects of microglial activation and in- Therefore, microglia display functional features of synaptic part- flammation have been studied (14, 23, 24), early consequences of ners, but their involvement in the regulation of synaptic trans- such activation are still unknown, especially the cell type involved mission has not yet been addressed. -

Human Ipsc-Derived Microglia Assume a Primary Microglia-Like State After Transplantation Into the Neonatal Mouse Brain
Human iPSC-derived microglia assume a primary microglia-like state after transplantation into the neonatal mouse brain Devon S. Svobodaa, M. Inmaculada Barrasaa,b, Jian Shua,c, Rosalie Rietjensa, Shupei Zhanga, Maya Mitalipovaa, Peter Berubec, Dongdong Fua, Leonard D. Shultzd, George W. Bella,b, and Rudolf Jaenischa,e,1 aWhitehead Institute for Biomedical Research, Cambridge, MA 02142; bBioinformatics and Research Computing, Whitehead Institute for Biomedical Research, Cambridge, MA 02142; cBroad Institute of MIT and Harvard, Cambridge, MA 02142; dThe Jackson Laboratory Cancer Center, The Jackson Laboratory, Bar Harbor, ME 04609; and eDepartment of Biology, Massachusetts Institute of Technology, Cambridge, MA 02142 Contributed by Rudolf Jaenisch, October 16, 2019 (sent for review August 8, 2019; reviewed by Valentina Fossati and Helmut Kettenmann) Microglia are essential for maintenance of normal brain function, Despite these innovations, there are important limitations to with dysregulation contributing to numerous neurological dis- modeling human disease with hiPSC-derived iMGs. As immune eases. Protocols have been developed to derive microglia-like cells cells, microglia are prone to activation and highly sensitive to from human induced pluripotent stem cells (hiPSCs). However, in vitro culture, which introduces impediments in extending re- primary microglia display major differences in morphology and sults obtained with cultured cells to disease states. This is high- gene expression when grown in culture, including down- lighted by recent studies showing that primary microglia directly regulation of signature microglial genes. Thus, in vitro differenti- isolated from the brain exhibit significant changes in gene ex- ated microglia may not accurately represent resting primary pression when grown in culture for as little as 6 h (26, 27). -

The Effects of Normal Aging on Myelin and Nerve Fibers: a Review∗
Journal of Neurocytology 31, 581–593 (2002) The effects of normal aging on myelin and nerve fibers: A review∗ ALAN PETERS Department of Anatomy and Neurobiology, Boston University School of Medicine, 715, Albany Street, Boston, MA 02118, USA [email protected] Received 7 January 2003; revised 4 March 2003; accepted 5 March 2003 Abstract It was believed that the cause of the cognitive decline exhibited by human and non-human primates during normal aging was a loss of cortical neurons. It is now known that significant numbers of cortical neurons are not lost and other bases for the cognitive decline have been sought. One contributing factor may be changes in nerve fibers. With age some myelin sheaths exhibit degenerative changes, such as the formation of splits containing electron dense cytoplasm, and the formation on myelin balloons. It is suggested that such degenerative changes lead to cognitive decline because they cause changes in conduction velocity, resulting in a disruption of the normal timing in neuronal circuits. Yet as degeneration occurs, other changes, such as the formation of redundant myelin and increasing thickness suggest of sheaths, suggest some myelin formation is continuing during aging. Another indication of this is that oligodendrocytes increase in number with age. In addition to the myelin changes, stereological studies have shown a loss of nerve fibers from the white matter of the cerebral hemispheres of humans, while other studies have shown a loss of nerve fibers from the optic nerves and anterior commissure in monkeys. It is likely that such nerve fiber loss also contributes to cognitive decline, because of the consequent decrease in connections between neurons.