Autophagy in Myelinating Glia
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

The National Economic Burden of Rare Disease Study February 2021
Acknowledgements This study was sponsored by the EveryLife Foundation for Rare Diseases and made possible through the collaborative efforts of the national rare disease community and key stakeholders. The EveryLife Foundation thanks all those who shared their expertise and insights to provide invaluable input to the study including: the Lewin Group, the EveryLife Community Congress membership, the Technical Advisory Group for this study, leadership from the National Center for Advancing Translational Sciences (NCATS) at the National Institutes of Health (NIH), the Undiagnosed Diseases Network (UDN), the Little Hercules Foundation, the Rare Disease Legislative Advocates (RDLA) Advisory Committee, SmithSolve, and our study funders. Most especially, we thank the members of our rare disease patient and caregiver community who participated in this effort and have helped to transform their lived experience into quantifiable data. LEWIN GROUP PROJECT STAFF Grace Yang, MPA, MA, Vice President Inna Cintina, PhD, Senior Consultant Matt Zhou, BS, Research Consultant Daniel Emont, MPH, Research Consultant Janice Lin, BS, Consultant Samuel Kallman, BA, BS, Research Consultant EVERYLIFE FOUNDATION PROJECT STAFF Annie Kennedy, BS, Chief of Policy and Advocacy Julia Jenkins, BA, Executive Director Jamie Sullivan, MPH, Director of Policy TECHNICAL ADVISORY GROUP Annie Kennedy, BS, Chief of Policy & Advocacy, EveryLife Foundation for Rare Diseases Anne Pariser, MD, Director, Office of Rare Diseases Research, National Center for Advancing Translational Sciences (NCATS), National Institutes of Health Elisabeth M. Oehrlein, PhD, MS, Senior Director, Research and Programs, National Health Council Christina Hartman, Senior Director of Advocacy, The Assistance Fund Kathleen Stratton, National Academies of Science, Engineering and Medicine (NASEM) Steve Silvestri, Director, Government Affairs, Neurocrine Biosciences Inc. -

Neuregulin 1–Erbb2 Signaling Is Required for the Establishment of Radial Glia and Their Transformation Into Astrocytes in Cerebral Cortex
Neuregulin 1–erbB2 signaling is required for the establishment of radial glia and their transformation into astrocytes in cerebral cortex Ralf S. Schmid*, Barbara McGrath*, Bridget E. Berechid†, Becky Boyles*, Mark Marchionni‡, Nenad Sˇ estan†, and Eva S. Anton*§ *University of North Carolina Neuroscience Center and Department of Cell and Molecular Physiology, University of North Carolina School of Medicine, Chapel Hill, NC 27599; †Department of Neurobiology, Yale University School of Medicine, New Haven, CT 06510; and ‡CeNes Pharamceuticals, Inc., Norwood, MA 02062 Communicated by Pasko Rakic, Yale University School of Medicine, New Haven, CT, January 27, 2003 (received for review December 12, 2002) Radial glial cells and astrocytes function to support the construction mine whether NRG-1-mediated signaling is involved in radial and maintenance, respectively, of the cerebral cortex. However, the glial cell development and differentiation in the cerebral cortex. mechanisms that determine how radial glial cells are established, We show that NRG-1 signaling, involving erbB2, may act in maintained, and transformed into astrocytes in the cerebral cortex are concert with Notch signaling to exert a critical influence in the not well understood. Here, we show that neuregulin-1 (NRG-1) exerts establishment, maintenance, and appropriate transformation of a critical role in the establishment of radial glial cells. Radial glial cell radial glial cells in cerebral cortex. generation is significantly impaired in NRG mutants, and this defect can be rescued by exogenous NRG-1. Down-regulation of expression Materials and Methods and activity of erbB2, a member of the NRG-1 receptor complex, leads Clonal Analysis to Study NRG’s Role in the Initial Establishment of to the transformation of radial glial cells into astrocytes. -

The Interplay Between Neurons and Glia in Synapse Development And
Available online at www.sciencedirect.com ScienceDirect The interplay between neurons and glia in synapse development and plasticity Jeff A Stogsdill and Cagla Eroglu In the brain, the formation of complex neuronal networks and regulate distinct aspects of synaptic development and amenable to experience-dependent remodeling is complicated circuit connectivity. by the diversity of neurons and synapse types. The establishment of a functional brain depends not only on The intricate communication between neurons and glia neurons, but also non-neuronal glial cells. Glia are in and their cooperative roles in synapse formation are now continuous bi-directional communication with neurons to direct coming to light due in large part to advances in genetic the formation and refinement of synaptic connectivity. This and imaging tools. This article will examine the progress article reviews important findings, which uncovered cellular made in our understanding of the role of mammalian and molecular aspects of the neuron–glia cross-talk that perisynaptic glia (astrocytes and microglia) in synapse govern the formation and remodeling of synapses and circuits. development, maturation, and plasticity since the previ- In vivo evidence demonstrating the critical interplay between ous Current Opinion article [1]. An integration of past and neurons and glia will be the major focus. Additional attention new findings of glial control of synapse development and will be given to how aberrant communication between neurons plasticity is tabulated in Box 1. and glia may contribute to neural pathologies. Address Glia control the formation of synaptic circuits Department of Cell Biology, Duke University Medical Center, Durham, In the CNS, glial cells are in tight association with NC 27710, USA synapses in all brain regions [2]. -

Autosomal Dominant Leukodystrophy with Autonomic Symptoms
List of Papers This thesis is based on the following papers, which are referred to in the text by their Roman numerals. I MR imaging characteristics and neuropathology of the spin- al cord in adult-onset autosomal dominant leukodystrophy with autonomic symptoms. Sundblom J, Melberg A, Kalimo H, Smits A, Raininko R. AJNR Am J Neuroradiol. 2009 Feb;30(2):328-35. II Genomic duplications mediate overexpression of lamin B1 in adult-onset autosomal dominant leukodystrophy (ADLD) with autonomic symptoms. Schuster J, Sundblom J, Thuresson AC, Hassin-Baer S, Klopstock T, Dichgans M, Cohen OS, Raininko R, Melberg A, Dahl N. Neurogenetics. 2011 Feb;12(1):65-72. III Bedside diagnosis of rippling muscle disease in CAV3 p.A46T mutation carriers. Sundblom J, Stålberg E, Osterdahl M, Rücker F, Montelius M, Kalimo H, Nennesmo I, Islander G, Smits A, Dahl N, Melberg A. Muscle Nerve. 2010 Jun;41(6):751-7. IV A family with discordance between Malignant hyperthermia susceptibility and Rippling muscle disease. Sundblom J, Mel- berg A, Rücker F, Smits A, Islander G. Manuscript submitted to Journal of Anesthesia. Reprints were made with permission from the respective publishers. Contents Introduction..................................................................................................11 Background and history............................................................................... 13 Early studies of hereditary disease...........................................................13 Darwin and Mendel................................................................................ -

Myelin Biogenesis Is Associated with Pathological Ultrastructure That Is
bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. 1 Myelin biogenesis is associated with pathological ultrastructure that 2 is resolved by microglia during development 3 4 5 Minou Djannatian1,2*, Ulrich Weikert3, Shima Safaiyan1,2, Christoph Wrede4, Cassandra 6 Deichsel1,2, Georg Kislinger1,2, Torben Ruhwedel3, Douglas S. Campbell5, Tjakko van Ham6, 7 Bettina Schmid2, Jan Hegermann4, Wiebke Möbius3, Martina Schifferer2,7, Mikael Simons1,2,7* 8 9 1Institute of Neuronal Cell Biology, Technical University Munich, 80802 Munich, Germany 10 2German Center for Neurodegenerative Diseases (DZNE), 81377 Munich, Germany 11 3Max-Planck Institute of Experimental Medicine, 37075 Göttingen, Germany 12 4Institute of Functional and Applied Anatomy, Research Core Unit Electron Microscopy, 13 Hannover Medical School, 30625, Hannover, Germany 14 5Department of Neuronal Remodeling, Graduate School of Pharmaceutical Sciences, Kyoto 15 University, Sakyo-ku, Kyoto 606-8501, Japan. 16 6Department of Clinical Genetics, Erasmus MC, University Medical Center Rotterdam, 3015 CN, 17 the Netherlands 18 7Munich Cluster of Systems Neurology (SyNergy), 81377 Munich, Germany 19 20 *Correspondence: [email protected] or [email protected] 21 Keywords 22 Myelination, degeneration, phagocytosis, microglia, oligodendrocytes, phosphatidylserine 23 24 1 bioRxiv preprint doi: https://doi.org/10.1101/2021.02.02.429485; this version posted February 4, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
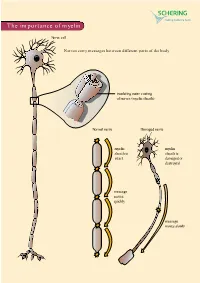
The Importance of Myelin
The importance of myelin Nerve cell Nerves carry messages between different parts of the body insulating outer coating of nerves (myelin sheath) Normal nerve Damaged nerve myelin myelin sheath is sheath is intact damaged or destroyed message moves quickly message moves slowly Nerve cells transmit impulses Nerve cells have a long, thin, flexible fibre that transmits impulses. These impulses are electrical signals that travel along the length of the nerve. Nerve fibres are long to enable impulses to travel between distant parts of the body, such as the spinal cord and leg muscles. Myelin speeds up impulses Most nerve fibres are surrounded by an insulating, fatty sheath called myelin, which acts to speed up impulses. The myelin sheath contains periodic breaks called nodes of Ranvier. By jumping from node to node, the impulse can travel much more quickly than if it had to travel along the entire length of the nerve fibre. Myelinated nerves can transmit a signal at speeds as high as 100 metres per second – as fast as a Formula One racing car. normal damaged nerve nerve Loss of myelin leads to a variety of symptoms If the myelin sheath surrounding nerve fibres is damaged or destroyed, transmission of nerve impulses is slowed or blocked. The impulse now has to flow continuously along the whole nerve fibre – a process that is much slower than jumping from node to node. Loss of myelin can also lead to ‘short-circuiting’ of nerve impulses. An area where myelin has been destroyed is called a lesion or plaque. This slowing and ‘short-circuiting’ of nerve impulses by lesions leads to a variety of symptoms related to nervous system activity. -

Regulation of Myelin Structure and Conduction Velocity by Perinodal Astrocytes
Correction NEUROSCIENCE Correction for “Regulation of myelin structure and conduc- tion velocity by perinodal astrocytes,” by Dipankar J. Dutta, Dong Ho Woo, Philip R. Lee, Sinisa Pajevic, Olena Bukalo, William C. Huffman, Hiroaki Wake, Peter J. Basser, Shahriar SheikhBahaei, Vanja Lazarevic, Jeffrey C. Smith, and R. Douglas Fields, which was first published October 29, 2018; 10.1073/ pnas.1811013115 (Proc. Natl. Acad. Sci. U.S.A. 115,11832–11837). The authors note that the following statement should be added to the Acknowledgments: “We acknowledge Dr. Hae Ung Lee for preliminary experiments that informed the ultimate experimental approach.” Published under the PNAS license. Published online June 10, 2019. www.pnas.org/cgi/doi/10.1073/pnas.1908361116 12574 | PNAS | June 18, 2019 | vol. 116 | no. 25 www.pnas.org Downloaded by guest on October 2, 2021 Regulation of myelin structure and conduction velocity by perinodal astrocytes Dipankar J. Duttaa,b, Dong Ho Wooa, Philip R. Leea, Sinisa Pajevicc, Olena Bukaloa, William C. Huffmana, Hiroaki Wakea, Peter J. Basserd, Shahriar SheikhBahaeie, Vanja Lazarevicf, Jeffrey C. Smithe, and R. Douglas Fieldsa,1 aSection on Nervous System Development and Plasticity, The Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892; bThe Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., Bethesda, MD 20817; cMathematical and Statistical Computing Laboratory, Office of Intramural Research, Center for Information -
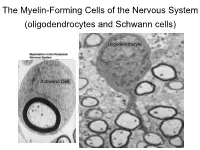
The Myelin-Forming Cells of the Nervous System (Oligodendrocytes and Schwann Cells)
The Myelin-Forming Cells of the Nervous System (oligodendrocytes and Schwann cells) Oligodendrocyte Schwann Cell Oligodendrocyte function Saltatory (jumping) nerve conduction Oligodendroglia PMD PMD Saltatory (jumping) nerve conduction Investigating the Myelinogenic Potential of Individual Oligodendrocytes In Vivo Sparse Labeling of Oligodendrocytes CNPase-GFP Variegated expression under the MBP-enhancer Cerebral Cortex Corpus Callosum Cerebral Cortex Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Corpus Callosum Cerebral Cortex Caudate Putamen Corpus Callosum Ant Commissure Corpus Callosum Cerebral Cortex Caudate Putamen Piriform Cortex Corpus Callosum Ant Commissure Characterization of Oligodendrocyte Morphology Cerebral Cortex Corpus Callosum Caudate Putamen Cerebellum Brain Stem Spinal Cord Oligodendrocytes in disease: Cerebral Palsy ! CP major cause of chronic neurological morbidity and mortality in children ! CP incidence now about 3/1000 live births compared to 1/1000 in 1980 when we started intervening for ELBW ! Of all ELBW {gestation 6 mo, Wt. 0.5kg} , 10-15% develop CP ! Prematurely born children prone to white matter injury {WMI}, principle reason for the increase in incidence of CP ! ! 12 Cerebral Palsy Spectrum of white matter injury ! ! Macro Cystic Micro Cystic Gliotic Khwaja and Volpe 2009 13 Rationale for Repair/Remyelination in Multiple Sclerosis Oligodendrocyte specification oligodendrocytes specified from the pMN after MNs - a ventral source of oligodendrocytes -

Myelin Oligodendrocyte Glycoprotein (MOG) Antibody Disease
MOG ANTIBODY DISEASE Myelin Oligodendrocyte Glycoprotein (MOG) Antibody-Associated Encephalomyelitis WHAT IS MOG ANTIBODY-ASSOCIATED DEMYELINATION? Myelin oligodendrocyte glycoprotein (MOG) antibody-associated demyelination is an immune-mediated inflammatory process that affects the central nervous system (brain and spinal cord). MOG is a protein that exists on the outer surface of cells that create myelin (an insulating layer around nerve fibers). In a small number of patients, an initial episode of inflammation due to MOG antibodies may be the first manifestation of multiple sclerosis (MS). Most patients may only experience one episode of inflammation, but repeated episodes of central nervous system demyelination can occur in some cases. WHAT ARE THE SYMPTOMS? • Optic neuritis (inflammation of the optic nerve(s)) may be a symptom of MOG antibody- associated demyelination, which may result in painful loss of vision in one or both eyes. • Transverse myelitis (inflammation of the spinal cord) may cause a variety of symptoms that include: o Abnormal sensations. Numbness, tingling, coldness or burning in the arms and/or legs. Some are especially sensitive to the light touch of clothing or to extreme heat or cold. You may feel as if something is tightly wrapping the skin of your chest, abdomen or legs. o Weakness in your arms or legs. Some people notice that they're stumbling or dragging one foot, or heaviness in the legs. Others may develop severe weakness or even total paralysis. o Bladder and bowel problems. This may include needing to urinate more frequently, urinary incontinence, difficulty urinating and constipation. • Acute disseminated encephalomyelitis (ADEM) may cause loss of vision, weakness, numbness, and loss of balance, and altered mental status. -

Follow-Up Study of 22 Chinese Children with Alexander Disease and Analysis of Parental Origin of De Novo GFAP Mutations
Journal of Human Genetics (2013) 58, 183–188 & 2013 The Japan Society of Human Genetics All rights reserved 1434-5161/13 www.nature.com/jhg ORIGINAL ARTICLE Follow-up study of 22 Chinese children with Alexander disease and analysis of parental origin of de novo GFAP mutations Lili Zang1, Jingmin Wang1, Yuwu Jiang1, Qiang Gu1, Zhijie Gao1, Yanling Yang1, Jiangxi Xiao2 and Ye Wu1 To delineate the phenotype and genotype in Chinese children with type I Alexander disease (AxD) and the parental origin of de novo glial fibrillary acidic protein (GFAP) mutations. Twenty-two children with clinically diagnosed type I AxD were followed up for 1.66–6.62 years. Allele-specific PCR was used for the analysis of parental origin of the allele harboring the de novo mutation. Phenotype of these patients were consistent with type I AxD described in other population, with developmental delay (motor delay in 81.82%, cognitive delay in 63.64%), macrocephaly (100%), seizures (95.45%), paroxysmal deterioration (27.27%) and typical brain magnetic resonance imaging (100%). Progression was slower than reported. At 8.55 years of age (5.29–13.25), all patients who underwent the second follow-up were alive. Eleven heterozygous missense mutations of GFAP were identified in 21 patients, with three novel mutations. Reported hot spot mutations, p.R79, p.R239 and p.R88, were also identified in Chinese patients. Mutations were de novo in all but one case. The mother of a proband was demonstrated to be a presymptomatic patient with type II AxD with a p.R79H mutation. Ninety percent of de novo mutations were on the paternal allele demonstrated by allele-specific PCR. -

Infantile Alexander Disease: Case Report and Review of Literature Dentistry Section
Case Report DOI: 10.7860/JCDR/2017/26875.10106 Infantile Alexander Disease: Case Report and Review of Literature Dentistry Section SOUMYABRATA SARKAR1, RUPAM SINHA2, AMITABHA CHAKRABORTY3, TANYA KHAITAN4, BIYAS BHOWMIK5 ABSTRACT Alexander Disease (AD) is an autosomal dominant leukodystrophy and occurs predominantly in infants and children. It usually results in death within ten years after onset. Among the four subtypes, infantile form comprises the most of affected individuals. It presents in the first two years of life, typically with progressive psychomotor deficiency, loss of developmental milestones, seizures, and pyramidal signs. Clinical and magnetic resonance image findings usually establish diagnosis of AD. Here, we present a case of Infantile AD with characteristic clinical and radiological features. Keywords: Leukodystrophy, Magnetic resonance imaging, Seizures CASE REPORT A five-year-old boy reported to the Department of Oral Medicine and Radiology, with painful decayed tooth in the upper right back jaw region for 10 days. The pain was mild, continuous, non- radiating in nature with no associated factors. The patient’s parents gave history of delayed developmental milestones of their son. There was no history of consanguineous marriage of the patient’s parents. They reported their son to be normal at birth with standard birth weight and head circumference. There was history of seizures six months after birth, with gradual psychomotor deficiencies and cognitive abnormalities, for which his parents sought paediatric consultation immediately at seven months of age. Initial consultation was inconclusive due to lack of diagnostic evidence and financial constrictions. However, he was put on phenytoin oral suspension twice daily, which controlled the symptoms. -

Neuron-Satellite Glial Cell Interactions in Sympathetic Nervous System Development
NEURON-SATELLITE GLIAL CELL INTERACTIONS IN SYMPATHETIC NERVOUS SYSTEM DEVELOPMENT by Erica D. Boehm A dissertation submitted to the Johns Hopkins University in conformity with the requirements for the degree of Doctor of Philosophy Baltimore, Maryland July 2020 © 2020 Erica Boehm All rights reserved. ABSTRACT Glial cells play crucial roles in maintaining the stability and structure of the nervous system. Satellite glial cells are a loosely defined population of glial cells that ensheathe neuronal cell bodies, dendrites, and synapses of the peripheral nervous system (Elfvin and Forsman 1978; Pannese 1981). Satellite glial cells are closely juxtaposed to peripheral neurons with only 20nm of space between their membranes (Dixon 1969). This close association suggests a tight coupling between the cells to allow for possible exchange of important nutrients, yet very little is known about satellite glial cell function and development. How neurons and glial cells co-develop to create this tightly knit unit remains undefined, as well as the functional consequences of disrupting these contacts. Satellite glial cells are derived from the same population of cells that give rise to peripheral neurons, but do not begin differentiation and proliferation until neurogenesis has been completed (Hall and Landis 1992). A key signaling pathway involved in glial specification is the Delta/Notch signaling pathway (Tsarovina et al. 2008). However, recent studies also implicate Notch signaling in the maturation of glia through non- canonical Notch ligands such as Delta/Notch-like EGF-related Receptor (DNER) (Eiraku et al. 2005). Interestingly, it has been reported that levels of DNER in sympathetic neurons may be dependent on the target-derived growth factor, nerve growth factor (NGF), and this signal is prominent in sympathetic neurons at the time in which satellite glial cells are developing (Deppmann et al.