Use of High-Throughput Sequencing for the Characterization of Extracellular RNA and to Study the Dynamics of Bacterial RNA Modification
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Intrinsically Disordered Proteins As Crucial Constituents of Cellular Aqueous Two Phase Systems and Coacervates ⇑ Vladimir N
View metadata, citation and similar papers at core.ac.uk brought to you by CORE provided by Elsevier - Publisher Connector FEBS Letters 589 (2015) 15–22 journal homepage: www.FEBSLetters.org Hypothesis Intrinsically disordered proteins as crucial constituents of cellular aqueous two phase systems and coacervates ⇑ Vladimir N. Uversky a,b,c,d, , Irina M. Kuznetsova d,e, Konstantin K. Turoverov d,e, Boris Zaslavsky f a Department of Molecular Medicine and USF Health Byrd Alzheimer’s Research Institute, Morsani College of Medicine, University of South Florida, Tampa, FL, USA b Institute for Biological Instrumentation, Russian Academy of Sciences, Pushchino, Moscow Region, Russian Federation c Biology Department, Faculty of Science, King Abdulaziz University, P.O. Box 80203, Jeddah 21589, Saudi Arabia d Laboratory of Structural Dynamics, Stability and Folding of Proteins, Institute of Cytology, Russian Academy of Sciences, St. Petersburg, Russian Federation e St. Petersburg State Polytechnical University, St. Petersburg, Russian Federation f AnalizaDx Inc., 3615 Superior Ave., Suite 4407B, Cleveland, OH 44114, USA article info abstract Article history: Here, we hypothesize that intrinsically disordered proteins (IDPs) serve as important drivers of the Received 17 August 2014 intracellular liquid–liquid phase separations that generate various membrane-less organelles. This Revised 10 October 2014 hypothesis is supported by the overwhelming abundance of IDPs in these organelles. Assembly and Accepted 19 November 2014 disassembly of these organelles are controlled by changes in the concentrations of IDPs, their post- Available online 29 November 2014 translational modifications, binding of specific partners, and changes in the pH and/or temperature Edited by A. Valencia of the solution. -

Dissertation Inês Silva ITQB.Pdf
The Role of Small RNAs and Ribonucleases in the Control of Gene Expression in Salmonella Typhimurium Inês de Jesus de Almeida e Silva Insert here an image with rounded corners Dissertation presented to obtain the Ph.D degree in Biology Instituto de Tecnologia Química e Biológica | Universidade Nova de Lisboa Oeiras, December, 2012 The Role of Small RNAs and Ribonucleases in the Control of Gene Expression in Salmonella Typhimurium Inês de Jesus de Almeida e Silva Dissertation presented to obtain the Ph.D degree in Biology Instituto de Tecnologia Química e Biológica Universidade Nova de Lisboa Oeiras, December, 2012 Financial Support from Fundação para a Ciência e Tecnologia (FCT) – Ph.D: grant - SFRH / BD / 43211 / 2008. Work performed at: Control of Gene Expression Laboratory Instituto de Tecnologia Química e Biológica Av. da República (EAN) 2781-901 Oeiras – Portugal Tel: +351-21-4469548 Fax: +351-21-4469549 Supervisor : Professora Doutora Cecília Maria Pais de Faria de Andrade Arraiano – Investigadora Coordenadora, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa. (Head of the Laboratory of Control of Gene Expression, where the work of this Dissertation was performed) Co-supervisor : Doutora Sandra Cristina de Oliveira Viegas – Investigadora Pós-Doutorada, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa. (Post-doc Fellow in the Laboratory of Control of Gene Expression, where the work of this Dissertation was performed) President of the Jury : Doutora Claudina Amélia Marques Rodrigues Pousada – Professora Catedrática Convidada do Instituto de Tecnologia Química e Biológica da Universidade Nova de Lisboa, por delegação; iii Examiners: Professor Doutor Iñigo Lasa Uzcudun – Head of Microbial Biofilm Research Group, Instituto de Agrobiotecnología, Pamplona (Principal Examiner). -
Generated by SRI International Pathway Tools Version 25.0, Authors S
An online version of this diagram is available at BioCyc.org. Biosynthetic pathways are positioned in the left of the cytoplasm, degradative pathways on the right, and reactions not assigned to any pathway are in the far right of the cytoplasm. Transporters and membrane proteins are shown on the membrane. Periplasmic (where appropriate) and extracellular reactions and proteins may also be shown. Pathways are colored according to their cellular function. Gcf_005222025Cyc: Escherichia coli 121 Cellular Overview Connections between pathways are omitted for legibility. pro a dicarboxylate a dicarboxylate an amino putrescine putrescine thiosulfate a hexose 6- glycine betaine glycine succinate γ-butyrobetaine L-ascorbate acid a dipeptide a dipeptide a dipeptide poly-β-1,6- 1-(β-D O-acetyl-L-serine O-acetyl-L-serine spermidine spermidine D-mannitol galactitol sulfate phosphate phosphate phosphate betaine Fe 3+ an amino an amino D-cellobiose an aromatic an aromatic a dipeptide 6 phosphate phosphate N-acetyl-D- thiamine cys cys spermidine spermidine protoheme 2+ citrate N -(D-fructosyl)-L-lysine L-proline betaine glutamate L-proline betaine L-carnitine acid an amino acid an amino an amino an amino an amino a tripeptide a tripeptide a tripeptide amino acid amino acid ribofuranosyl) (R)- putrescine Fe N-acetyl-D- molybdate phosphate a hydroxamate 3+ 3+ arbutin shikimate enterobactin 2-oxoglutarate N 6 -(D-psicosyl)-L-lysine D-sorbitol D-glucosamine D-mannitol galactitol N-acetylneuraminate N-acetylneuraminate sulfate sulfate sulfate glycine betaine -
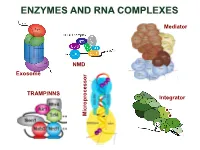
Enzymes and Rna Complexes
ENZYMES AND RNA COMPLEXES Mediator NMD Exosome NMD TRAMP/NNS Integrator Microprocessor RNA PROCESSING and DECAY machinery: RNases Protein Function Characteristics Exonucleases 5’ 3’ Xrn1 cytoplasmic, mRNA degradation processsive Rat1 nuclear, pre-rRNA, sn/snoRNA, pre-mRNA processing and degradation Rrp17/hNol12 nuclear, pre-rRNA processing Exosome 3’ 5’ multisubunit exo/endo complex subunits organized as in bacterial PNPase Rrp44/Dis3 catalytic subunit Exo/PIN domains, processsive Rrp4, Rrp40 pre-rRNA, sn/snoRNA processing, mRNA degradation Rrp41-43, 45-46 participates in NMD, ARE-dependent, non-stop decay Mtr3, Ski4 Mtr4 nuclear helicase cofactor DEAD box Rrp6 (Rrp47) nuclear exonuclease ( Rrp6 BP, cofactor) RNAse D homolog, processsive Ski2,3,7,8 cytoplasmic exosome cofactors. SKI complex helicase, GTPase Other 3’ 5’ Rex1-4 3’-5’ exonucleases, rRNA, snoRNA, tRNA processing RNase D homolog DXO 3’-5’ exonuclease in addition to decapping mtEXO 3’ 5’ mitochondrial degradosome RNA degradation in yeast Suv3/ Dss1 helicase/ 3’-5’ exonuclease DExH box/ RNase II homolog Deadenylation Ccr4/NOT/Pop2 major deadenylase complex (Ccr, Caf, Pop, Not proteins) Ccr4- Mg2+ dependent endonuclease Pan2p/Pan3 additional deadenylases (poliA tail length) RNase D homolog, poly(A) specific nuclease PARN mammalian deadenylase RNase D homolog, poly(A) specific nuclease Endonucleases RNase III -Rnt1 pre-rRNA, sn/snoRNA processing, mRNA degradation dsRNA specific -Dicer, Drosha siRNA/miRNA biogenesis, functions in RNAi PAZ, RNA BD, RNase III domains Ago2 Slicer -

SUPLEMENTARY MATERIAL 1) Comparative Analysis of The
1 SUPLEMENTARY MATERIAL 2 1) Comparative analysis of the different differential expression methods applied to the 3 transcriptome of Vanilla planifolia Jacks. 4 5 6 7 8 9 10 11 12 13 14 2dpi 10dpi 15 16 Supplemental Figure S1. Venn diagram showing the comparison of the differentially 17 expressed unigenes obtained with the methods DESeq2, EdgeR, NOISeq, and DESeq. At the 18 center of the diagram we observed that the EdgeR method comprises the great majority of 19 genes determined by the other methods. The right panel corresponds to 2 dpi, while the left 20 panel corresponds to 10 dpi. 21 22 23 24 25 26 2) Global expression profiles in response to infection caused by Fusarium 27 oxysporum f. sp. vanillae in vanilla. 28 . 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 2dpi 10dpi 51 Supplemental Figure S2. Heat map that contrasts the global vanilla response to 52 Fusarium oxysporum f. sp. vanillae. On the right we observe the early response 53 (2dpi); while in the left panel it presents the response to 10dpi. All differentially 54 expressed unigenes are included. 55 56 3) Expression profiles related to biotic stress, in the late response (10dpi) of 57 vanilla to Fusarium oxysporum f. sp. vanillae 58 59 60 61 62 63 64 65 66 67 68 69 70 71 Supplemental Figure S3. Heat map indicating the expression profiles of the annotated DEG 72 unigenes, corresponding to 10dpi. The numbers in the figure correspond to different 73 categories of gene ontology, as described below: 25 C1-metabolism, 11 lipid metabolism, 3 74 minor CHO metabolism, 13 amino acid metabolism, 16 secondary metabolism, 26 misc, 17 75 hormone metabolism, 30 signalling, 31 cell, 23 nucleotide metabolism, 27 RNA, 28 DNA, 76 33 development, 24 Biodegradation of Xenobiotics, 18 Co-factor and vitamine metabolism, 77 35 not assigned, 34 transport, 29 protein, 20 stress, 2 major CHO metabolism, 10 cell wall. -

Ribonuclease A
Chem. Rev. 1998, 98, 1045−1065 1045 Ribonuclease A Ronald T. Raines Departments of Biochemistry and Chemistry, University of WisconsinsMadison, Madison, Wisconsin 53706 Received October 10, 1997 (Revised Manuscript Received January 12, 1998) Contents I. Introduction 1045 II. Heterologous Production 1046 III. Structure 1046 IV. Folding and Stability 1047 A. Disulfide Bond Formation 1047 B. Prolyl Peptide Bond Isomerization 1048 V. RNA Binding 1048 A. Subsites 1048 B. Substrate Specificity 1049 C. One-Dimensional Diffusion 1049 D. Processive Catalysis 1050 VI. Substrates 1050 VII. Inhibitors 1051 Ronald T. Raines was born in 1958 in Montclair, NJ. He received Sc.B. VIII. Reaction Mechanism 1052 degrees in chemistry and biology from the Massachusetts Institute of A. His12 and His119 1053 Technology. At M.I.T., he worked with Christopher T. Walsh to reveal the reaction mechanisms of pyridoxal 5′-phosphate-dependent enzymes. B. Lys41 1054 Raines was a National Institutes of Health predoctoral fellow in the C. Asp121 1055 chemistry department at Harvard University. There, he worked with D. Gln11 1056 Jeremy R. Knowles to elucidate the reaction energetics of triosephosphate IX. Reaction Energetics 1056 isomerase. Raines was a Helen Hay Whitney postdoctoral fellow in the biochemistry and biophysics department at the University of California, A. Transphosphorylation versus Hydrolysis 1056 San Francisco. At U.C.S.F., he worked with William J. Rutter to clone, B. Rate Enhancement 1057 express, and mutate the cDNA that codes for ribonuclease A. Raines X. Ribonuclease S 1058 then joined the faculty of the biochemistry department at the University s A. S-Protein−S-Peptide Interaction 1058 of Wisconin Madison, where he is now associate professor of biochem- istry and chemistry. -

Ribonuclease E Organizes the Protein Interactions in the Escherichia Coli RNA Degradosome
Downloaded from genesdev.cshlp.org on September 26, 2021 - Published by Cold Spring Harbor Laboratory Press Ribonuclease E organizes the protein interactions in the Escherichia coli RNA degradosome Nathalie F. Vanzo,1 Yeun Shan Li,2 Be´atrice Py,2,3 Erwin Blum,2 Christopher F. Higgins,2,4 Lelia C. Raynal,1 Henry M. Krisch,1 and Agamemnon J. Carpousis1,5 1Laboratoire de Microbiologie et Ge´ne´tique Mole´culaire, UPR 9007, Centre National de la Recherche Scientifique (CNRS), 31062 Toulouse Cedex, France; 2Nuffield Department of Clinical Biochemistry and Imperial Cancer Research Fund Laboratories, Institute of Molecular Medicine, John Radcliffe Hospital, University of Oxford, Oxford OX3 9DS, UK The Escherichia coli RNA degradosome is the prototype of a recently discovered family of multiprotein machines involved in the processing and degradation of RNA. The interactions between the various protein components of the RNA degradosome were investigated by Far Western blotting, the yeast two-hybrid assay, and coimmunopurification experiments. Our results demonstrate that the carboxy-terminal half (CTH) of ribonuclease E (RNase E) contains the binding sites for the three other major degradosomal components, the DEAD-box RNA helicase RhlB, enolase, and polynucleotide phosphorylase (PNPase). The CTH of RNase E acts as the scaffold of the complex upon which the other degradosomal components are assembled. Regions for oligomerization were detected in the amino-terminal and central regions of RNase E. Furthermore, polypeptides derived from the highly charged region of RNase E, containing the RhlB binding site, stimulate RhlB activity at least 15-fold, saturating at one polypeptide per RhlB molecule. A model for the regulation of the RhlB RNA helicase activity is presented. -

Characterization of the Mammalian RNA Exonuclease 5/NEF-Sp As a Testis-Specific Nuclear 3′′′′′ → 5′′′′′ Exoribonuclease
Downloaded from rnajournal.cshlp.org on October 7, 2021 - Published by Cold Spring Harbor Laboratory Press Characterization of the mammalian RNA exonuclease 5/NEF-sp as a testis-specific nuclear 3′′′′′ → 5′′′′′ exoribonuclease SARA SILVA,1,2 DAVID HOMOLKA,1 and RAMESH S. PILLAI1 1Department of Molecular Biology, University of Geneva, CH-1211 Geneva 4, Switzerland 2European Molecular Biology Laboratory, Grenoble Outstation, 38042, France ABSTRACT Ribonucleases catalyze maturation of functional RNAs or mediate degradation of cellular transcripts, activities that are critical for gene expression control. Here we identify a previously uncharacterized mammalian nuclease family member NEF-sp (RNA exonuclease 5 [REXO5] or LOC81691) as a testis-specific factor. Recombinant human NEF-sp demonstrates a divalent metal ion-dependent 3′′′′′ → 5′′′′′ exoribonuclease activity. This activity is specific to single-stranded RNA substrates and is independent of their length. The presence of a 2′′′′′-O-methyl modification at the 3′′′′′ end of the RNA substrate is inhibitory. Ectopically expressed NEF-sp localizes to the nucleolar/nuclear compartment in mammalian cell cultures and this is dependent on an amino-terminal nuclear localization signal. Finally, mice lacking NEF-sp are viable and display normal fertility, likely indicating overlapping functions with other nucleases. Taken together, our study provides the first biochemical and genetic exploration of the role of the NEF-sp exoribonuclease in the mammalian genome. Keywords: NEF-sp; LOC81691; Q96IC2; REXON; RNA exonuclease 5; REXO5; 2610020H08Rik INTRODUCTION clease-mediated processing to create their final 3′ ends: poly(A) tails of most mRNAs or the hairpin structure of Spermatogenesis is the process by which sperm cells are replication-dependent histone mRNAs (Colgan and Manley produced in the male germline. -

Protein Engineering to Exploit and Explore Bovine Secretory Ribonucleases
PROTEIN ENGINEERING TO EXPLOIT A N D EXPLORE BOVINE SECRETORY RIBONUCLEASES by JIN-SOO KIM A thesis submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy (Biochemistry) at the UNIVERSITY OF WISCONSIN-MADISON 1994 Reproduced with permission of the copyright owner. Further reproduction prohibited without permission. ACKNOWLEDGEMENTS I would like to thank Dr. Ronald T. Raines for his advice and support. His scientific insight has been very helpful throughout this work. I would also like to thank the entire Raines group for their friendship and companionship. I am grateful to Dr. J. Soucek and Dr. J. Matousek for their collaboration with us, which has been a valuable part of the BS-RNase research. I thank Dr. M. Karpeisky for suggesting the protein fusion project, and Dr. G. D'Alessio and Dr. L. Mazzarella for providing the coordinates of BS-RNase. I have been generously supported by Steenbock predoctoral fellowship from the Department of Biochemistry. Finally, I thank my parents, who have encouraged (or at least not discouraged) me to pursue a career in science since I was a kid. Reproduced with permission of the copyright owner. Further reproduction prohibited without permission ABSTRACT PROTEIN ENGINEERING TO EXPLOIT AND EXPLORE BOVINE SECRETORY RIBONUCLEASES Jin-Soo Kim Under the supervision of Dr. Ronald T. Raines at the University of Wisconsin-Madison Ribonuclease S-peptide (residues 1-20) and S-protein (residues 21- 124) are the enzymatically inactive products of the limited digestion of bovine pancreatic ribonuclease A (RNase A) by subtilisin. S-Peptide binds S-protein with high affinity to form RNase S, which has full enzymatic activity. -

Rnase E Activity Is Conferred by a Single Polypeptide
Proc. Natl. Acad. Sci. USA Vol. 90, pp. 9006-9010, October 1993 Biochemustry RNase E activity is conferred by a single polypeptide: Overexpression, purification, and properties of the ams/rne/hmpl gene product (endoribonuclease/RNA processing) R. S. CORMACK, J. L. GENEREAUX, AND G. A. MACKIE* Department of Biochemistry, University of Western Ontario, London, Ontario, Canada N6A 5C1 Communicated by Myron K. Brakke, June 29, 1993 (receivedfor review May 10, 1993) ABSTRACT Ribonuclease E, an enzyme that processes encoding a high molecular mass protein hypothesized to be pre-SS rRNA from Its precursor, is now believed to be the involved in nucleoid partitioning or cell wall invagination major endoribonuclease participating in mRNA turnover in during division (16). The revised DNA sequence, coterminal Escherichia coli. The product of the ams/rne/hmpl gene, with the 5' end of the two previous sequences, encodes a which is required for RNase E activity, was overexpressed, protein of 1025 amino acid residues whose predicted relative purified to near homogeneity by electroelution from an molecular mass would be 114,000. This polypeptide displays SDS/polyacrylamide gel, and renatured. The purified poly- anomalous electrophoretic mobility in SDS/polyacrylamide peptide possesses nucleolytic activity in vitro with a specificity gels, however, and migrates with an apparent size of 180 kDa identical to that observed for crude RNase E preparations. In (16). It has not been proven that the polypeptide encoded by addition, both UV crosslinking and RNA-protein blotting the ams/rne/hmpl gene is, in fact, a ribonuclease or is unambiguously showed that the Ams/Rne/Hmpl polypeptide intimately associated with the cleavage ofRNA either in vitro has a high affnity for RNA. -

Supplementary Information
Supplementary information (a) (b) Figure S1. Resistant (a) and sensitive (b) gene scores plotted against subsystems involved in cell regulation. The small circles represent the individual hits and the large circles represent the mean of each subsystem. Each individual score signifies the mean of 12 trials – three biological and four technical. The p-value was calculated as a two-tailed t-test and significance was determined using the Benjamini-Hochberg procedure; false discovery rate was selected to be 0.1. Plots constructed using Pathway Tools, Omics Dashboard. Figure S2. Connectivity map displaying the predicted functional associations between the silver-resistant gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S3. Connectivity map displaying the predicted functional associations between the silver-sensitive gene hits; disconnected gene hits not shown. The thicknesses of the lines indicate the degree of confidence prediction for the given interaction, based on fusion, co-occurrence, experimental and co-expression data. Figure produced using STRING (version 10.5) and a medium confidence score (approximate probability) of 0.4. Figure S4. Metabolic overview of the pathways in Escherichia coli. The pathways involved in silver-resistance are coloured according to respective normalized score. Each individual score represents the mean of 12 trials – three biological and four technical. Amino acid – upward pointing triangle, carbohydrate – square, proteins – diamond, purines – vertical ellipse, cofactor – downward pointing triangle, tRNA – tee, and other – circle. -

Supplementary Information
Electronic Supplementary Material (ESI) for ChemComm. This journal is © The Royal Society of Chemistry 2020 Supplementary Information Electrical tension-triggered conversion of anaerobic to aerobic respiration of Shewanella putrefaciens CN32 while promoting biofilm growth in microbial fuel cells Xiu He a, Xiaoshuai Wu c,Yan Qiao a*,Tianbao Hu a, Deng Wang a, Xiao Han a and Changt Ming Li a ,b ,c ,d* a Institute for Clean Energy & Advanced Materials, Faculty of Materials & Energy, Southwest University, No. 2 T iansheng Road, Chongqing 400715, P. R. China. b Chongqing Engineering Research Center for Rapid Diagnosis of Dread Disease, Southwest University, Chongqin g 400715, P. R. China. c Institute of Materials Science and Devices, Suzhou University of Science and Technology, Suzhou 215011, P. R. China. d Institute of Advanced Crossfield Science, Qingdao University, Qingdao 266071, P. R. China. * Corresponding author. E-mail: yanqiao@ swu.edu.cn; [email protected] Experimental section: Bacteria culture A single colony of S. putrefaciens CN32 (ATCC, BBA-1097) was cultured aerobically in 100 ml LB at 30 °C overnight1 and the cells were harvested and inoculated into fresh LB. After grown at 30 °C for 6 hours, the cells were by centrifuging at 4 °C (6000 rpm, 5 min) for MFC half-cell operation2. Before every test, nitrogen was purged into the suspension for 30 min to remove oxygen from the cell. MFC half-cell operation For the anodic half-cell analysis, a piece of 1 cm∗1cm a NiO nanowire decelerated carbon cloth was used as working electrode, saturated calomel electrode (SCE) was used as the reference electrode and titanium plate as the counter electrode3.