202834Orig1s000
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CHLORZOXAZONE- Chlorzoxazone Tablet DIRECT RX ------CHLORZOXAZONE
CHLORZOXAZONE- chlorzoxazone tablet DIRECT RX ---------- CHLORZOXAZONE DESCRIPTION SECTION Chlorzoxazone USP is a centrally acting skeletal muscle relaxant, available as tablets of 500 mg for oral administration. Its chemical name is 5-Chloro-2-benzoxazolinone, and its structural formula is: C7H4CINO2 MW 169.57 Chlorzoxazone USP is a white or practically white, practically odorless, crystalline powder. Chlorzoxazone is slightly soluble in water; sparingly soluble in alcohol, in isopropyl alcohol, and in methanol; soluble in solutions of alkali hydroxides and ammonia. Chlorzoxazone tablets contain the inactive ingredients Docusate Sodium, Lactose (hydrous), Magnesium Stearate, Microcrystalline Cellulose, Pregelatinized Starch, Sodium Benzoate, and Sodium Starch Glycolate. CLINICAL PHARMACOLOGY SECTION Chlorzoxazone is a centrally-acting agent for painful musculoskeletal conditions. Data available from animal experiments as well as human study indicate that chlorzoxazone acts primarily at the level of the spinal cord and subcortical areas of the brain where it inhibits multisynaptic reflex arcs involved in producing and maintaining skeletal muscle spasm of varied etiology. The clinical result is a reduction of the skeletal muscle spasm with relief of pain and increased mobility of the involved muscles. Blood levels of chlorzoxazone can be detected in people during the first 30 minutes and peak levels may be reached, in the majority of the subjects, in about 1 to 2 hours after oral administration of chlorzoxazone. Chlorzoxazone is rapidly metabolized and is excreted in the urine, primarily in a conjugated form as the glucuronide. Less than one percent of a dose of chlorzoxazone is excreted unchanged in the urine in 24 hours. INDICATIONS & USAGE SECTION Chlorzoxazone is indicated as an adjunct to rest, physical therapy, and other measures for the relief of discomfort associated with acute, painful musculoskeletal conditions. -

LORZONE- Chlorzoxazone Tablet Vertical Pharmaceuticals , LLC ---For Painful Musculoskeletal Conditions PRESCRIBING INFOR
LORZONE- chlorzoxazone tablet Vertical Pharmaceuticals , LLC ---------- For Painful Musculoskeletal Conditions PRESCRIBING INFORMATION DESCRIPTION Each 375 mg Lorzone® tablet contains: chlorzoxazone USP 375 mg. Each 750 mg Lorzone® tablet contains: chlorzoxazone USP 750 mg. Chemical Name: 5-Chloro-2-benzoxazolinone. Structural Formula: Molecular Formula: C7H4CINO2 Molecular Weight: 169.56 Chlorzoxazone USP is a white or practically white, practically odorless, crystalline powder. Chlorzoxazone is slightly soluble in water; sparingly soluble in alcohol, in isopropyl alcohol, and in methanol; soluble in solutions of alkali hydroxides and ammonia. Inactive ingredients: anhydrous lactose, croscarmellose sodium, docusate sodium, magnesium stearate, microcrystalline cellulose, pregelatinized corn starch and sodium benzoate. CLINICAL PHARMACOLOGY Chlorzoxazone is a centrally-acting agent for painful musculoskeletal conditions. Data available from animal experiments as well as human study indicate that chlorzoxazone acts primarily at the level of the spinal cord and subcortical areas of the brain where it inhibits multisynaptic reflex arcs involved in producing and maintaining skeletal muscle spasm of varied etiology. The clinical result is a reduction of the skeletal muscle spasm with relief of pain and increased mobility of the involved muscles. Blood levels of chlorzoxazone can be detected in people during the first 30 minutes and peak levels may be reached, in the majority of the subjects, in about 1 to 2 hours after oral administration of chlorzoxazone. Chlorzoxazone is rapidly metabolized and is excreted in the urine, primarily in a conjugated form as the glucuronide. Less than one percent of a dose of chlorzoxazone is excreted unchanged in the urine in 24 hours. INDICATIONS AND USAGE Lorzone® is indicated as an adjunct to rest, physical therapy, and other measures for the relief of discomfort associated with acute, painful musculoskeletal conditions. -

Drug Class Review on Skeletal Muscle Relaxants
Drug Class Review on Skeletal Muscle Relaxants Final Report Update 2 May 2005 Original Report Date: April 2003 Update 1 Report Date: January 2004 A literature scan of this topic is done periodically The purpose of this report is to make available information regarding the comparative effectiveness and safety profiles of different drugs within pharmaceutical classes. Reports are not usage guidelines, nor should they be read as an endorsement of, or recommendation for, any particular drug, use or approach. Oregon Health & Science University does not recommend or endorse any guideline or recommendation developed by users of these reports. Roger Chou, MD Kim Peterson, MS Oregon Evidence-based Practice Center Oregon Health & Science University Mark Helfand, MD, MPH, Director Copyright © 2005 by Oregon Health & Science University Portland, Oregon 97201. All rights reserved. Note: A scan of the medical literature relating to the topic is done periodically (see http://www.ohsu.edu/ohsuedu/research/policycenter/DERP/about/methods.cfm for scanning process description). Upon review of the last scan, the Drug Effectiveness Review Project governance group elected not to proceed with another full update of this report. Some portions of the report may not be up to date. Prior versions of this report can be accessed at the DERP website. Final Report Update 2 Drug Effectiveness Review Project TABLE OF CONTENTS Introduction........................................................................................................................4 Scope and -

San Juan County Adult Drug Court Participant Handbook
Participant Manual SEVENTH DISTRICT ADULT DRUG COURT MONTICELLO, UTAH Updated January 2018 Subject to Change 1 2 Welcome to the San Juan County Adult Drug Court This Handbook is designed to introduce you to the San Juan County Drug Court program, answer your questions and provide overall information about the Drug Court Program. As a participant, you will be expected to follow the instructions given in Drug Court by the Judge and comply with the treatment plan developed for you by the treatment team. If you are reading this Handbook it means that we are confident that Drug Court will help you to learn how to make successful choices free of the influence of drugs or alcohol. 3 Table of Contents Welcome to the San Juan County Adult Drug Court ...................................................................... 3 Overview ....................................................................................................................................... 6 Drug Court Team ........................................................................................................................... 7 Judge’s Role ........................................................................................................................... 7 San Juan County Attorney’s Role (Prosecutor) ....................................................................... 8 Defense Attorney Role (Your Attorney) ................................................................................... 8 Probation Officer’s Role ......................................................................................................... -

Pharmaceutical Appendix to the Harmonized Tariff Schedule
Harmonized Tariff Schedule of the United States Basic Revision 3 (2021) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States Basic Revision 3 (2021) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names INN which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service CAS registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. -

Original Research Article Comparison of Different Brands of Centrally Acting Skeletal Muscle Relaxants: a Cost Analysis Study
Print ISSN: 2319-2003 | Online ISSN: 2279-0780 IJBCP International Journal of Basic & Clinical Pharmacology DOI: http://dx.doi.org/10.18203/2319-2003.ijbcp20192213 Original Research Article Comparison of different brands of centrally acting skeletal muscle relaxants: a cost analysis study Kiran B., Kala P.*, Chitra N. S., Jamuna Rani R.* Department of Pharmacology, ABSTRACT SRM Medical College and Research Centre, Background: Skeletal muscle relaxants are structurally distinct drugs prescribed Kattankulathur, for reducing muscle spasms, pain, and hyperreflexia. Centrally acting skeletal Kancheepuram, Tamil Nadu, muscle relaxants are manufactured by various pharmaceutical companies with India variable price. The present study, aimed to analyze the cost variation of various brands of centrally acting skeletal muscle relaxants, so as to help the physician to Received: 15 April 2019 choose the cost effective treatment. Accepted: 13 May 2019 Methods: Current index of medical stores (CIMS) April 2018 and online literature were used as information guide to review the prices of drugs used in the *Correspondence to: treatment of musculo skeletal pain and spastic neurological disorders. Dr. Kala P., Results: Among anti spasmodic group, thiocolchicoside 4 mg shows maximum Email: kalaramesh75@ price variation of 337.5%, whereas carisoprodol 350 mg shows the least variation gmail.com of 0.1%. It is evident from antispastic group that baclofen 10 mg shows maximum price variation of 93.91% and 5 mg of Baclofen shows the least variation of Copyright: © the author(s), 11.22%. It is observed that, among anti spastic group, a percentage prize variation publisher and licensee Medip of 93.91 for 10 mg and 11.22 for 5 mg baclofen. -

Eagling Differential Selectivity of CYP Inhibitors Against Probe Substrates
Br J Clin Pharmacol 1998; 45: 107–114 Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes Victoria A. Eagling, John F. Tjia & David J. Back Department of Pharmacology & Therapeutics, University of Liverpool, P.O. Box 147, Liverpool L69 3GE, UK Aims Chemical inhibitors of cytochrome P450 (CYP) are a useful tool in defining the role of individual CYPs involved in drug metabolism. The aim of the present study was to evaluate the selectivity and rank the order of potency of a range of isoform-selective CYP inhibitors and to compare directly the eVects of these inhibitors in human and rat hepatic microsomes. Methods Four chemical inhibitors of human cytochrome P450 isoforms, furafylline (CYP1A2), sulphaphenazole (CYP2C9), diethyldithiocarbamate (CYP2E1), and ketoconazole (CYP3A4) were screened for their inhibitory specificity towards CYP- mediated reactions in both human and rat liver microsomal preparations. Phenacetin O-deethylation, tolbutamide 4-hydroxylation, chlorzoxazone 6-hydroxylation and testosterone 6b-hydroxylation were monitored for enzyme activity. Results Furafylline was a potent, selective inhibitor of phenacetin O-deethylation = (CYP1A2-mediated) in human liver microsomes (IC50 0.48 mm), but inhibited both phenacetin O-deethylation and tolbutamide 4-hydroxylation (CYP2C9- mediated) at equimolar concentrations in rat liver microsomes (IC50=20.8 and 24.0 mm respectively). Sulphaphenazole demonstrated selective inhibition of tolbutamide hydroxylation in human liver microsomes but failed to inhibit this reaction in rat liver microsomes. DDC demonstrated a low level of selectivity as an inhibitory probe for chlorzoxazone 6-hydroxylation (CYP2E1-mediated). DDC also inhibited testosterone 6b-hydroxylation (CYP3A-mediated) in man and rat, and tolbutamide 4-hydroxylase activity in rat. -
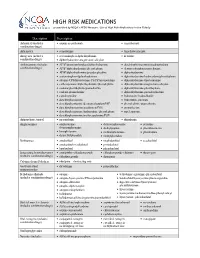
HIGH RISK MEDICATIONS As Specified by NCQA’S HEDIS Measure: Use of High Risk Medications in the Elderly
HIGH RISK MEDICATIONS as specified by NCQA’s HEDIS Measure: Use of High Risk Medications in the Elderly Description Prescription Antianxiety (includes • aspirin-meprobamate • meprobamate combination drugs) Antiemetics • scopolamine • trimethobenzamide Analgesics (includes • acetaminophen-diphenhydramine • ketorolac combination drugs) • diphenhydramine-magnesium salicylate Antihistamines (includes • APAP/dextromethorphan/diphenhydramine • dexchlorpheniramine-pseudoephedrine combination drugs) • APAP/diphenhydramine/phenylephrine • dextromethorphan-promethazine • APAP/diphenhydramine/pseudoephedrine • diphenhydramine • acetaminophen-diphenhydramine • diphenhydramine/hydrocodone/phenylephrine • atropine/CPM/hyoscyamine/PE/PPA/scopolamine • diphenhydramine-tripelennamine • carbetapentane/diphenhydramine/phenylephrine • diphenhydramine-magnesium salicylate • codeine/phenylephrine/promethazine • diphenhydramine-phenylephrine • codeine-promethazine • diphenhydramine-pseudoephedrine • cyproheptadine • hydroxyzine hydrochloride • dexchlorpheniramine • hydroxyzine pamoate • dexchlorpheniramine/dextromethorphan/PSE • phenylephrine-promethazine • dexchlorpheniramine/guaifenesin/PSE • promethazine • dexchlorpheniramine/hydrocodone/phenylephrine • tripelennamine • dexchlorpheniramine/methscopolamine/PSE Antipsychotic, typical • mesoridazine • thioridazine Amphetamines • amphetamine- • dextroamphetamine • pemoline dextroamphetamine • diethylpropion • phendimetrazine • benzphetamine • methamphetamine • phentermine • dexmethylphenidate • methylphenidate -

United States Patent (19) 11 Patent Number: 4,780,463 Sunshine Et Al
United States Patent (19) 11 Patent Number: 4,780,463 Sunshine et al. (45) Date of Patent: Oct. 25, 1988 (54). ANALGESIC, ANTI-INFLAMMATORY AND Ailments of the Musculoskeletal Apparatus), Investiga SKELETAL MUSCLE RELAXANT tion Medica Internactional, pp. 475-478, (1983), and COMPOSITIONS COMPRISING English translation thereof. NON-STEROIDAL ANTI-NFLAMMATORY Socialist Republic of Romania Description of Inven DRUGS AND MUSCULOSKELETAL tion, 82,717, copy of patent and English translation RELAXANTS AND METHODS OF USING thereof. SAME Rego, "Mio-Relaxantes No Tratamento Das Lom 75 Inventors: Abraham Sunshine, New York; balgias Aguda E Da Lombo-Ciaticas Recentes', Mus Eugene M. Laska, Larchmont; cle Relaxants in the Treatment of Acute Lumbalgias Carole E. Siegel, Mamaroneck, all of and Recent Lumbo-Sciatica Cases, Acta N.Y. Reumatologica Portuguesa, II, 2:363-364, (1974), copy of the original and English translation thereof. 73 Assignee: Analgesic Associates, Larchmont, Schror, "Analgetisch-antiphlogistische Therapie Von N.Y. Schmerzzustanden des Bewegungsapparates', Anal 21 Appl. No.: 114,751. gesic-Antiphlogistic Therapy of Locomotor System Pain), Therapiewoche, 28, 5657-5663, (1978), copy of the 22 Filed: Oct. 30, 1987 original and English translation thereof. Schar, "Medikamentose Behandling von Lumboishial Related U.S. Application Data gien', Drug Treatment of the Lumbago-Sciatic Syn 60 Division of Ser. No. 815,502, Jan. 2, 1986, Pat. No. drome, Schweiz. Rundschau Med., (Praxis), vol. 68, No. 4,722,938, which is a continuation of Ser. No. 686,380, 5, pp. 141-142, (Jan. 30, 1979), copy of original article Dec. 26, 1984, abandoned. and English translation thereof. 51) int. Cl.' ..................... A61K 31/19; A61K 31/44; Kolodny and Klipper, "Bone and Joint Diseases in the A61K 3/54; A61K 31/195; A61K 31/205 Elderly', Hospital Practice, pp. -

Lorzone (Chlorzoxazone) Dose, Indications, Adverse Effects, Interactions... from PDR.Net 19/7/17 0:18
Lorzone (chlorzoxazone) dose, indications, adverse effects, interactions... from PDR.net 19/7/17 0:18 print Close window CLASSES Muscle Relaxants, Centrally Acting, Plain DEA CLASS Rx DESCRIPTION Oral, centrally acting, skeletal muscle relaxant; use has fallen out of favor; drug has been associated with hepatotoxicity. COMMON BRAND NAMES Lorzone, Parafon Forte DSC, Relax-DS HOW SUPPLIED Chlorzoxazone/Lorzone/Parafon Forte DSC/Relax-DS Oral Tab: 375mg, 500mg, 750mg DOSAGE & INDICATIONS For adjunctive therapy to rest, physical therapy, and other measures for the relief of musculoskeletal pain associated with acute, painful musculoskeletal conditions. Oral dosage Adults, Adolescents, and the Geriatric Initially, 250 to 500 mg PO given 3 to 4 times per day. Doses up to 750 mg PO given 3 to 4 times per day may be given for severe muscle spasm, but reduce when possible to lowest effective dose. Use with extreme caution in geriatric patients, as unpredictable, fatal hepatotoxicity has been reported. Children† Initially, 20 mg/kg/day PO given in 3 to 4 divided doses; or 600 mg/m2/day PO given in 3 to 4 divided doses. MAXIMUM DOSAGE Adults 3000 mg/day PO. Elderly 3000 mg/day PO. Adolescents 3000 mg/day PO. Children Maximum dosage limits are not available. DOSING CONSIDERATIONS Hepatic Impairment Avoid use in patients with hepatic impairment. Although causal factors are not known, chlorzoxazone has been associated with severe (and fatal) hepatotoxicity, even in patients without concurrent hepatic impairment. Renal Impairment No dosage adjustment is recommended; however, use with caution as metabolites are excreted via the kidneys. ADMINISTRATION Oral Administration Oral Solid Formulations If stomach upset occurs, chlorzoxazone may be taken with food or milk. -

The Relative Impact of Solubility and Mobility From
Stability of Amorphous Solid Dispersions – The Relative Impact of Solubility and Mobility from Molecular Dynamics Simulations — Supporting Information Michael Brunsteiner,† Johannes Khinast,†,‡ and Amrit Paudel∗,†,‡ Research Center Pharmaceutical Engineering, Graz, Austria, and University of Technology, Graz, Austria E-mail: [email protected] ∗To whom correspondence should be addressed †Research Center Pharmaceutical Engineering, Graz, Austria ‡University of Technology, Graz, Austria 1 1 Choice of Model Systems Lidocaine (LI) Benzamide (BD) Phenacetin (PH) Bifonazole (BI) Chlorzoxazone (CZ) Flufenamic acid (FFA) Flurbiprofen (FB) Chlorpropamide (CP) Figure S1: Literature data1 for eight different API molecules, each formulated with four different polymer types. ASD stabilities are given as amorphicity index (AI) values that are each an average from measurements at several different API loadings. 2 2 Convergence -100 -20 FLA PAC -101 -21 -102 -22 [kJ/mol] [kJ/mol] -103 -23 nonb nonb E E -104 -24 -105 -25 0 20 40 60 80 100 120 140 160 180 200 0 20 40 60 80 100 120 140 160 180 200 time [ns] time [ns] FLA PAC 0.354 0.274 0.352 0.272 ] ] 3 3 0.35 0.27 Volume [nm 0.348 Volume [nm 0.268 0.346 0.266 0 20 40 60 80 100 120 140 160 180 200 0 20 40 60 80 100 120 140 160 180 200 time [ns] time [ns] Figure S2: Convergence of simulations of pure APIs. Non-bonded energies (top) and volume per molecule (bottom). 20 0.4 FLA FLA PAC PAC 0 0.38 -20 0.36 ] 3 -40 0.34 [kJ/mol] -60 0.32 nonb Volume [nm E -80 0.3 -100 0.28 -120 0.26 0 20 40 60 80 100 120 140 160 180 200 0 20 40 60 80 100 120 140 160 180 200 time [ns] time [ns] Figure S3: Convergence of simulations of pure APIs. -

Skeletal Muscle Relaxants Therapeutic Class Review (TCR) December 3, 2012
Skeletal Muscle Relaxants Therapeutic Class Review (TCR) December 3, 2012 Please Note: This clinical document has been retired. It can be used as a historical reference. No part of this publication may be reproduced or transmitted in any form or by any means, electronic or mechanical, including photocopying, recording, digital scanning, or via any information storage or retrieval system without the express written consent of Magellan Rx Management. All requests for permission should be mailed to: Magellan Rx Management Attention: Legal Department 6950 Columbia Gateway Drive Columbia, Maryland 21046 The materials contained herein represent the opinions of the collective authors and editors and should not be construed to be the official representation of any professional organization or group, any state Pharmacy and Therapeutics committee, any state Medicaid Agency, or any other clinical committee. This material is not intended to be relied upon as medical advice for specific medical cases and nothing contained herein should be relied upon by any patient, medical professional or layperson seeking information about a specific course of treatment for a specific medical condition. All readers of this material are responsible for independently obtaining medical advice and guidance from their own physician and/or other medical professional in regard to the best course of treatment for their specific medical condition. This publication, inclusive of all forms contained herein, is intended to be educational in nature and is intended to be used for informational purposes only. Send comments and suggestions to [email protected]. Proprietary Information. Restricted Access – Do not disseminate or copy without approval. © 2007–2012 Magellan Rx Management.