Optimalization of Nanosuspension
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

CHLORZOXAZONE- Chlorzoxazone Tablet DIRECT RX ------CHLORZOXAZONE
CHLORZOXAZONE- chlorzoxazone tablet DIRECT RX ---------- CHLORZOXAZONE DESCRIPTION SECTION Chlorzoxazone USP is a centrally acting skeletal muscle relaxant, available as tablets of 500 mg for oral administration. Its chemical name is 5-Chloro-2-benzoxazolinone, and its structural formula is: C7H4CINO2 MW 169.57 Chlorzoxazone USP is a white or practically white, practically odorless, crystalline powder. Chlorzoxazone is slightly soluble in water; sparingly soluble in alcohol, in isopropyl alcohol, and in methanol; soluble in solutions of alkali hydroxides and ammonia. Chlorzoxazone tablets contain the inactive ingredients Docusate Sodium, Lactose (hydrous), Magnesium Stearate, Microcrystalline Cellulose, Pregelatinized Starch, Sodium Benzoate, and Sodium Starch Glycolate. CLINICAL PHARMACOLOGY SECTION Chlorzoxazone is a centrally-acting agent for painful musculoskeletal conditions. Data available from animal experiments as well as human study indicate that chlorzoxazone acts primarily at the level of the spinal cord and subcortical areas of the brain where it inhibits multisynaptic reflex arcs involved in producing and maintaining skeletal muscle spasm of varied etiology. The clinical result is a reduction of the skeletal muscle spasm with relief of pain and increased mobility of the involved muscles. Blood levels of chlorzoxazone can be detected in people during the first 30 minutes and peak levels may be reached, in the majority of the subjects, in about 1 to 2 hours after oral administration of chlorzoxazone. Chlorzoxazone is rapidly metabolized and is excreted in the urine, primarily in a conjugated form as the glucuronide. Less than one percent of a dose of chlorzoxazone is excreted unchanged in the urine in 24 hours. INDICATIONS & USAGE SECTION Chlorzoxazone is indicated as an adjunct to rest, physical therapy, and other measures for the relief of discomfort associated with acute, painful musculoskeletal conditions. -

LORZONE- Chlorzoxazone Tablet Vertical Pharmaceuticals , LLC ---For Painful Musculoskeletal Conditions PRESCRIBING INFOR
LORZONE- chlorzoxazone tablet Vertical Pharmaceuticals , LLC ---------- For Painful Musculoskeletal Conditions PRESCRIBING INFORMATION DESCRIPTION Each 375 mg Lorzone® tablet contains: chlorzoxazone USP 375 mg. Each 750 mg Lorzone® tablet contains: chlorzoxazone USP 750 mg. Chemical Name: 5-Chloro-2-benzoxazolinone. Structural Formula: Molecular Formula: C7H4CINO2 Molecular Weight: 169.56 Chlorzoxazone USP is a white or practically white, practically odorless, crystalline powder. Chlorzoxazone is slightly soluble in water; sparingly soluble in alcohol, in isopropyl alcohol, and in methanol; soluble in solutions of alkali hydroxides and ammonia. Inactive ingredients: anhydrous lactose, croscarmellose sodium, docusate sodium, magnesium stearate, microcrystalline cellulose, pregelatinized corn starch and sodium benzoate. CLINICAL PHARMACOLOGY Chlorzoxazone is a centrally-acting agent for painful musculoskeletal conditions. Data available from animal experiments as well as human study indicate that chlorzoxazone acts primarily at the level of the spinal cord and subcortical areas of the brain where it inhibits multisynaptic reflex arcs involved in producing and maintaining skeletal muscle spasm of varied etiology. The clinical result is a reduction of the skeletal muscle spasm with relief of pain and increased mobility of the involved muscles. Blood levels of chlorzoxazone can be detected in people during the first 30 minutes and peak levels may be reached, in the majority of the subjects, in about 1 to 2 hours after oral administration of chlorzoxazone. Chlorzoxazone is rapidly metabolized and is excreted in the urine, primarily in a conjugated form as the glucuronide. Less than one percent of a dose of chlorzoxazone is excreted unchanged in the urine in 24 hours. INDICATIONS AND USAGE Lorzone® is indicated as an adjunct to rest, physical therapy, and other measures for the relief of discomfort associated with acute, painful musculoskeletal conditions. -

Optum Essential Health Benefits Enhanced Formulary PDL January
PENICILLINS ketorolac tromethamineQL GENERIC mefenamic acid amoxicillin/clavulanate potassium nabumetone amoxicillin/clavulanate potassium ER naproxen January 2016 ampicillin naproxen sodium ampicillin sodium naproxen sodium CR ESSENTIAL HEALTH BENEFITS ampicillin-sulbactam naproxen sodium ER ENHANCED PREFERRED DRUG LIST nafcillin sodium naproxen DR The Optum Preferred Drug List is a guide identifying oxacillin sodium oxaprozin preferred brand-name medicines within select penicillin G potassium piroxicam therapeutic categories. The Preferred Drug List may piperacillin sodium/ tazobactam sulindac not include all drugs covered by your prescription sodium tolmetin sodium drug benefit. Generic medicines are available within many of the therapeutic categories listed, in addition piperacillin sodium/tazobactam Fenoprofen Calcium sodium to categories not listed, and should be considered Meclofenamate Sodium piperacillin/tazobactam as the first line of prescribing. Tolmetin Sodium Amoxicillin/Clavulanate Potassium LOW COST GENERIC PREFERRED For benefit coverage or restrictions please check indomethacin your benefit plan document(s). This listing is revised Augmentin meloxicam periodically as new drugs and new prescribing LOW COST GENERIC naproxen kit information becomes available. It is recommended amoxicillin that you bring this list of medications when you or a dicloxacillin sodium CARDIOVASCULAR covered family member sees a physician or other penicillin v potassium ACE-INHIBITORS healthcare provider. GENERIC QUINOLONES captopril ANTI-INFECTIVES -

DESCRIPTION Tolazamide Is an Oral Blood-Glucose-Lowering Drug of the Sulfonylurea Class
TOLAZAMIDE- tolazamide tablet PD-Rx Pharmaceuticals, Inc. ---------- DESCRIPTION Tolazamide is an oral blood-glucose-lowering drug of the sulfonylurea class. Tolazamide is a white or creamy-white powder very slightly soluble in water and slightly soluble in alcohol. The chemical name is 1-(Hexahydro-1 H-azepin-1-yl)-3-( p-tolylsulfonyl)urea. Tolazamide has the following structural formula: Each tablet for oral administration contains 250 mg or 500 mg of tolazamide, USP and the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and sodium lauryl sulfate. CLINICAL PHARMACOLOGY Actions Tolazamide appears to lower the blood glucose acutely by stimulating the release of insulin from the pancreas, an effect dependent upon functioning beta cells in the pancreatic islets. The mechanism by which tolazamide lowers blood glucose during long-term administration has not been clearly established. With chronic administration in type II diabetic patients, the blood glucose-lowering effect persists despite a gradual decline in the insulin secretory response to the drug. Extrapancreatic effects may be involved in the mechanism of action of oral sulfonylurea hypoglycemic drugs. Some patients who are initially responsive to oral hypoglycemic drugs, including tolazamide tablets, may become unresponsive or poorly responsive over time. Alternatively, tolazamide tablets may be effective in some patients who have become unresponsive to one or more other sulfonylurea drugs. In addition to its blood glucose-lowering actions, tolazamide produces a mild diuresis by enhancement of renal free water clearance. Pharmacokinetics Tolazamide is rapidly and well absorbed from the gastrointestinal tract. Peak serum concentrations occur at 3 to 4 hours following a single oral dose of the drug. -

Alcohol-Medication Interactions: the Acetaldehyde Syndrome
arm Ph ac f ov l o i a g n il r a n u c o e J Journal of Pharmacovigilance Borja-Oliveira, J Pharmacovigilance 2014, 2:5 ISSN: 2329-6887 DOI: 10.4172/2329-6887.1000145 Review Article Open Access Alcohol-Medication Interactions: The Acetaldehyde Syndrome Caroline R Borja-Oliveira* University of São Paulo, School of Arts, Sciences and Humanities, São Paulo 03828-000, Brazil *Corresponding author: Caroline R Borja-Oliveira, University of São Paulo, School of Arts, Sciences and Humanities, Av. Arlindo Bettio, 1000, Ermelino Matarazzo, São Paulo 03828-000, Brazil, Tel: +55-11-30911027; E-mail: [email protected] Received date: August 21, 2014, Accepted date: September 11, 2014, Published date: September 20, 2014 Copyright: © 2014 Borja-Oliveira CR. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited. Abstract Medications that inhibit aldehyde dehydrogenase when coadministered with alcohol produce accumulation of acetaldehyde. Acetaldehyde toxic effects are characterized by facial flushing, nausea, vomiting, tachycardia and hypotension, symptoms known as acetaldehyde syndrome, disulfiram-like reactions or antabuse effects. Severe and even fatal outcomes are reported. Besides the aversive drugs used in alcohol dependence disulfiram and cyanamide (carbimide), several other pharmaceutical agents are known to produce alcohol intolerance, such as certain anti-infectives, as cephalosporins, nitroimidazoles and furazolidone, dermatological preparations, as tacrolimus and pimecrolimus, as well as chlorpropamide and nilutamide. The reactions are also observed in some individuals after the simultaneous use of products containing alcohol and disulfiram-like reactions inducers. -

Sulfonylureas
Therapeutic Class Overview Sulfonylureas INTRODUCTION In the United States (US), diabetes mellitus affects more than 30 million people and is the 7th leading cause of death (Centers for Disease Control and Prevention [CDC] 2018). Type 2 diabetes mellitus (T2DM) is the most common form of diabetes and is characterized by elevated fasting and postprandial glucose concentrations (American Diabetes Association [ADA] 2019[a]). It is a chronic illness that requires continuing medical care and ongoing patient self-management education and support to prevent acute complications and to reduce the risk of long-term complications (ADA 2019[b]). ○ Complications of T2DM include hypertension, heart disease, stroke, vision loss, nephropathy, and neuropathy (ADA 2019[a]). In addition to dietary and lifestyle management, T2DM can be treated with insulin, one or more oral medications, or a combination of both. Many patients with T2DM will require combination therapy (Garber et al 2019). Classes of oral medications for the management of blood glucose levels in patients with T2DM focus on increasing insulin secretion, increasing insulin responsiveness, or both, decreasing the rate of carbohydrate absorption, decreasing the rate of hepatic glucose production, decreasing the rate of glucagon secretion, and blocking glucose reabsorption by the kidney (Garber et al 2019). Pharmacologic options for T2DM include sulfonylureas (SFUs), biguanides, thiazolidinediones (TZDs), meglitinides, alpha-glucosidase inhibitors, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagon-like peptide-1 (GLP-1) analogs, amylinomimetics, sodium-glucose cotransporter 2 (SGLT2) inhibitors, combination products, and insulin (Garber et al 2019). SFUs are the oldest of the oral antidiabetic medications, and all agents are available generically. The SFUs can be divided into 2 categories: first-generation and second-generation. -

Drug Class Review on Skeletal Muscle Relaxants
Drug Class Review on Skeletal Muscle Relaxants Final Report Update 2 May 2005 Original Report Date: April 2003 Update 1 Report Date: January 2004 A literature scan of this topic is done periodically The purpose of this report is to make available information regarding the comparative effectiveness and safety profiles of different drugs within pharmaceutical classes. Reports are not usage guidelines, nor should they be read as an endorsement of, or recommendation for, any particular drug, use or approach. Oregon Health & Science University does not recommend or endorse any guideline or recommendation developed by users of these reports. Roger Chou, MD Kim Peterson, MS Oregon Evidence-based Practice Center Oregon Health & Science University Mark Helfand, MD, MPH, Director Copyright © 2005 by Oregon Health & Science University Portland, Oregon 97201. All rights reserved. Note: A scan of the medical literature relating to the topic is done periodically (see http://www.ohsu.edu/ohsuedu/research/policycenter/DERP/about/methods.cfm for scanning process description). Upon review of the last scan, the Drug Effectiveness Review Project governance group elected not to proceed with another full update of this report. Some portions of the report may not be up to date. Prior versions of this report can be accessed at the DERP website. Final Report Update 2 Drug Effectiveness Review Project TABLE OF CONTENTS Introduction........................................................................................................................4 Scope and -

San Juan County Adult Drug Court Participant Handbook
Participant Manual SEVENTH DISTRICT ADULT DRUG COURT MONTICELLO, UTAH Updated January 2018 Subject to Change 1 2 Welcome to the San Juan County Adult Drug Court This Handbook is designed to introduce you to the San Juan County Drug Court program, answer your questions and provide overall information about the Drug Court Program. As a participant, you will be expected to follow the instructions given in Drug Court by the Judge and comply with the treatment plan developed for you by the treatment team. If you are reading this Handbook it means that we are confident that Drug Court will help you to learn how to make successful choices free of the influence of drugs or alcohol. 3 Table of Contents Welcome to the San Juan County Adult Drug Court ...................................................................... 3 Overview ....................................................................................................................................... 6 Drug Court Team ........................................................................................................................... 7 Judge’s Role ........................................................................................................................... 7 San Juan County Attorney’s Role (Prosecutor) ....................................................................... 8 Defense Attorney Role (Your Attorney) ................................................................................... 8 Probation Officer’s Role ......................................................................................................... -

Pharmaceutical Appendix to the Harmonized Tariff Schedule
Harmonized Tariff Schedule of the United States Basic Revision 3 (2021) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE HARMONIZED TARIFF SCHEDULE Harmonized Tariff Schedule of the United States Basic Revision 3 (2021) Annotated for Statistical Reporting Purposes PHARMACEUTICAL APPENDIX TO THE TARIFF SCHEDULE 2 Table 1. This table enumerates products described by International Non-proprietary Names INN which shall be entered free of duty under general note 13 to the tariff schedule. The Chemical Abstracts Service CAS registry numbers also set forth in this table are included to assist in the identification of the products concerned. For purposes of the tariff schedule, any references to a product enumerated in this table includes such product by whatever name known. -

Original Research Article Comparison of Different Brands of Centrally Acting Skeletal Muscle Relaxants: a Cost Analysis Study
Print ISSN: 2319-2003 | Online ISSN: 2279-0780 IJBCP International Journal of Basic & Clinical Pharmacology DOI: http://dx.doi.org/10.18203/2319-2003.ijbcp20192213 Original Research Article Comparison of different brands of centrally acting skeletal muscle relaxants: a cost analysis study Kiran B., Kala P.*, Chitra N. S., Jamuna Rani R.* Department of Pharmacology, ABSTRACT SRM Medical College and Research Centre, Background: Skeletal muscle relaxants are structurally distinct drugs prescribed Kattankulathur, for reducing muscle spasms, pain, and hyperreflexia. Centrally acting skeletal Kancheepuram, Tamil Nadu, muscle relaxants are manufactured by various pharmaceutical companies with India variable price. The present study, aimed to analyze the cost variation of various brands of centrally acting skeletal muscle relaxants, so as to help the physician to Received: 15 April 2019 choose the cost effective treatment. Accepted: 13 May 2019 Methods: Current index of medical stores (CIMS) April 2018 and online literature were used as information guide to review the prices of drugs used in the *Correspondence to: treatment of musculo skeletal pain and spastic neurological disorders. Dr. Kala P., Results: Among anti spasmodic group, thiocolchicoside 4 mg shows maximum Email: kalaramesh75@ price variation of 337.5%, whereas carisoprodol 350 mg shows the least variation gmail.com of 0.1%. It is evident from antispastic group that baclofen 10 mg shows maximum price variation of 93.91% and 5 mg of Baclofen shows the least variation of Copyright: © the author(s), 11.22%. It is observed that, among anti spastic group, a percentage prize variation publisher and licensee Medip of 93.91 for 10 mg and 11.22 for 5 mg baclofen. -

Eagling Differential Selectivity of CYP Inhibitors Against Probe Substrates
Br J Clin Pharmacol 1998; 45: 107–114 Differential selectivity of cytochrome P450 inhibitors against probe substrates in human and rat liver microsomes Victoria A. Eagling, John F. Tjia & David J. Back Department of Pharmacology & Therapeutics, University of Liverpool, P.O. Box 147, Liverpool L69 3GE, UK Aims Chemical inhibitors of cytochrome P450 (CYP) are a useful tool in defining the role of individual CYPs involved in drug metabolism. The aim of the present study was to evaluate the selectivity and rank the order of potency of a range of isoform-selective CYP inhibitors and to compare directly the eVects of these inhibitors in human and rat hepatic microsomes. Methods Four chemical inhibitors of human cytochrome P450 isoforms, furafylline (CYP1A2), sulphaphenazole (CYP2C9), diethyldithiocarbamate (CYP2E1), and ketoconazole (CYP3A4) were screened for their inhibitory specificity towards CYP- mediated reactions in both human and rat liver microsomal preparations. Phenacetin O-deethylation, tolbutamide 4-hydroxylation, chlorzoxazone 6-hydroxylation and testosterone 6b-hydroxylation were monitored for enzyme activity. Results Furafylline was a potent, selective inhibitor of phenacetin O-deethylation = (CYP1A2-mediated) in human liver microsomes (IC50 0.48 mm), but inhibited both phenacetin O-deethylation and tolbutamide 4-hydroxylation (CYP2C9- mediated) at equimolar concentrations in rat liver microsomes (IC50=20.8 and 24.0 mm respectively). Sulphaphenazole demonstrated selective inhibition of tolbutamide hydroxylation in human liver microsomes but failed to inhibit this reaction in rat liver microsomes. DDC demonstrated a low level of selectivity as an inhibitory probe for chlorzoxazone 6-hydroxylation (CYP2E1-mediated). DDC also inhibited testosterone 6b-hydroxylation (CYP3A-mediated) in man and rat, and tolbutamide 4-hydroxylase activity in rat. -
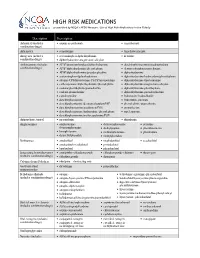
HIGH RISK MEDICATIONS As Specified by NCQA’S HEDIS Measure: Use of High Risk Medications in the Elderly
HIGH RISK MEDICATIONS as specified by NCQA’s HEDIS Measure: Use of High Risk Medications in the Elderly Description Prescription Antianxiety (includes • aspirin-meprobamate • meprobamate combination drugs) Antiemetics • scopolamine • trimethobenzamide Analgesics (includes • acetaminophen-diphenhydramine • ketorolac combination drugs) • diphenhydramine-magnesium salicylate Antihistamines (includes • APAP/dextromethorphan/diphenhydramine • dexchlorpheniramine-pseudoephedrine combination drugs) • APAP/diphenhydramine/phenylephrine • dextromethorphan-promethazine • APAP/diphenhydramine/pseudoephedrine • diphenhydramine • acetaminophen-diphenhydramine • diphenhydramine/hydrocodone/phenylephrine • atropine/CPM/hyoscyamine/PE/PPA/scopolamine • diphenhydramine-tripelennamine • carbetapentane/diphenhydramine/phenylephrine • diphenhydramine-magnesium salicylate • codeine/phenylephrine/promethazine • diphenhydramine-phenylephrine • codeine-promethazine • diphenhydramine-pseudoephedrine • cyproheptadine • hydroxyzine hydrochloride • dexchlorpheniramine • hydroxyzine pamoate • dexchlorpheniramine/dextromethorphan/PSE • phenylephrine-promethazine • dexchlorpheniramine/guaifenesin/PSE • promethazine • dexchlorpheniramine/hydrocodone/phenylephrine • tripelennamine • dexchlorpheniramine/methscopolamine/PSE Antipsychotic, typical • mesoridazine • thioridazine Amphetamines • amphetamine- • dextroamphetamine • pemoline dextroamphetamine • diethylpropion • phendimetrazine • benzphetamine • methamphetamine • phentermine • dexmethylphenidate • methylphenidate