The Role of Epigenetic Modifications, Long-Range Contacts, Enhancers
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

ARTICLE Doi:10.1038/Nature10523
ARTICLE doi:10.1038/nature10523 Spatio-temporal transcriptome of the human brain Hyo Jung Kang1*, Yuka Imamura Kawasawa1*, Feng Cheng1*, Ying Zhu1*, Xuming Xu1*, Mingfeng Li1*, Andre´ M. M. Sousa1,2, Mihovil Pletikos1,3, Kyle A. Meyer1, Goran Sedmak1,3, Tobias Guennel4, Yurae Shin1, Matthew B. Johnson1,Zˇeljka Krsnik1, Simone Mayer1,5, Sofia Fertuzinhos1, Sheila Umlauf6, Steven N. Lisgo7, Alexander Vortmeyer8, Daniel R. Weinberger9, Shrikant Mane6, Thomas M. Hyde9,10, Anita Huttner8, Mark Reimers4, Joel E. Kleinman9 & Nenad Sˇestan1 Brain development and function depend on the precise regulation of gene expression. However, our understanding of the complexity and dynamics of the transcriptome of the human brain is incomplete. Here we report the generation and analysis of exon-level transcriptome and associated genotyping data, representing males and females of different ethnicities, from multiple brain regions and neocortical areas of developing and adult post-mortem human brains. We found that 86 per cent of the genes analysed were expressed, and that 90 per cent of these were differentially regulated at the whole-transcript or exon level across brain regions and/or time. The majority of these spatio-temporal differences were detected before birth, with subsequent increases in the similarity among regional transcriptomes. The transcriptome is organized into distinct co-expression networks, and shows sex-biased gene expression and exon usage. We also profiled trajectories of genes associated with neurobiological categories and diseases, and identified associations between single nucleotide polymorphisms and gene expression. This study provides a comprehensive data set on the human brain transcriptome and insights into the transcriptional foundations of human neurodevelopment. -

Growth and Molecular Profile of Lung Cancer Cells Expressing Ectopic LKB1: Down-Regulation of the Phosphatidylinositol 3-Phosphate Kinase/PTEN Pathway1
[CANCER RESEARCH 63, 1382–1388, March 15, 2003] Growth and Molecular Profile of Lung Cancer Cells Expressing Ectopic LKB1: Down-Regulation of the Phosphatidylinositol 3-Phosphate Kinase/PTEN Pathway1 Ana I. Jimenez, Paloma Fernandez, Orlando Dominguez, Ana Dopazo, and Montserrat Sanchez-Cespedes2 Molecular Pathology Program [A. I. J., P. F., M. S-C.], Genomics Unit [O. D.], and Microarray Analysis Unit [A. D.], Spanish National Cancer Center, 28029 Madrid, Spain ABSTRACT the cell cycle in G1 (8, 9). However, the intrinsic mechanism by which LKB1 activity is regulated in cells and how it leads to the suppression Germ-line mutations in LKB1 gene cause the Peutz-Jeghers syndrome of cell growth is still unknown. It has been proposed that growth (PJS), a genetic disease with increased risk of malignancies. Recently, suppression by LKB1 is mediated through p21 in a p53-dependent LKB1-inactivating mutations have been identified in one-third of sporadic lung adenocarcinomas, indicating that LKB1 gene inactivation is critical in mechanism (7). In addition, it has been observed that LKB1 binds to tumors other than those of the PJS syndrome. However, the in vivo brahma-related gene 1 protein (BRG1) and this interaction is required substrates of LKB1 and its role in cancer development have not been for BRG1-induced growth arrest (10). Similar to what happens in the completely elucidated. Here we show that overexpression of wild-type PJS, Lkb1 heterozygous knockout mice show gastrointestinal hamar- LKB1 protein in A549 lung adenocarcinomas cells leads to cell-growth tomatous polyposis and frequent hepatocellular carcinomas (11, 12). suppression. To examine changes in gene expression profiles subsequent to Interestingly, the hamartomas, but not the malignant tumors, arising in exogenous wild-type LKB1 in A549 cells, we used cDNA microarrays. -

Supplementary Materials
Supplementary materials Supplementary Table S1: MGNC compound library Ingredien Molecule Caco- Mol ID MW AlogP OB (%) BBB DL FASA- HL t Name Name 2 shengdi MOL012254 campesterol 400.8 7.63 37.58 1.34 0.98 0.7 0.21 20.2 shengdi MOL000519 coniferin 314.4 3.16 31.11 0.42 -0.2 0.3 0.27 74.6 beta- shengdi MOL000359 414.8 8.08 36.91 1.32 0.99 0.8 0.23 20.2 sitosterol pachymic shengdi MOL000289 528.9 6.54 33.63 0.1 -0.6 0.8 0 9.27 acid Poricoic acid shengdi MOL000291 484.7 5.64 30.52 -0.08 -0.9 0.8 0 8.67 B Chrysanthem shengdi MOL004492 585 8.24 38.72 0.51 -1 0.6 0.3 17.5 axanthin 20- shengdi MOL011455 Hexadecano 418.6 1.91 32.7 -0.24 -0.4 0.7 0.29 104 ylingenol huanglian MOL001454 berberine 336.4 3.45 36.86 1.24 0.57 0.8 0.19 6.57 huanglian MOL013352 Obacunone 454.6 2.68 43.29 0.01 -0.4 0.8 0.31 -13 huanglian MOL002894 berberrubine 322.4 3.2 35.74 1.07 0.17 0.7 0.24 6.46 huanglian MOL002897 epiberberine 336.4 3.45 43.09 1.17 0.4 0.8 0.19 6.1 huanglian MOL002903 (R)-Canadine 339.4 3.4 55.37 1.04 0.57 0.8 0.2 6.41 huanglian MOL002904 Berlambine 351.4 2.49 36.68 0.97 0.17 0.8 0.28 7.33 Corchorosid huanglian MOL002907 404.6 1.34 105 -0.91 -1.3 0.8 0.29 6.68 e A_qt Magnogrand huanglian MOL000622 266.4 1.18 63.71 0.02 -0.2 0.2 0.3 3.17 iolide huanglian MOL000762 Palmidin A 510.5 4.52 35.36 -0.38 -1.5 0.7 0.39 33.2 huanglian MOL000785 palmatine 352.4 3.65 64.6 1.33 0.37 0.7 0.13 2.25 huanglian MOL000098 quercetin 302.3 1.5 46.43 0.05 -0.8 0.3 0.38 14.4 huanglian MOL001458 coptisine 320.3 3.25 30.67 1.21 0.32 0.9 0.26 9.33 huanglian MOL002668 Worenine -

Literature Mining Sustains and Enhances Knowledge Discovery from Omic Studies
LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES by Rick Matthew Jordan B.S. Biology, University of Pittsburgh, 1996 M.S. Molecular Biology/Biotechnology, East Carolina University, 2001 M.S. Biomedical Informatics, University of Pittsburgh, 2005 Submitted to the Graduate Faculty of School of Medicine in partial fulfillment of the requirements for the degree of Doctor of Philosophy University of Pittsburgh 2016 UNIVERSITY OF PITTSBURGH SCHOOL OF MEDICINE This dissertation was presented by Rick Matthew Jordan It was defended on December 2, 2015 and approved by Shyam Visweswaran, M.D., Ph.D., Associate Professor Rebecca Jacobson, M.D., M.S., Professor Songjian Lu, Ph.D., Assistant Professor Dissertation Advisor: Vanathi Gopalakrishnan, Ph.D., Associate Professor ii Copyright © by Rick Matthew Jordan 2016 iii LITERATURE MINING SUSTAINS AND ENHANCES KNOWLEDGE DISCOVERY FROM OMIC STUDIES Rick Matthew Jordan, M.S. University of Pittsburgh, 2016 Genomic, proteomic and other experimentally generated data from studies of biological systems aiming to discover disease biomarkers are currently analyzed without sufficient supporting evidence from the literature due to complexities associated with automated processing. Extracting prior knowledge about markers associated with biological sample types and disease states from the literature is tedious, and little research has been performed to understand how to use this knowledge to inform the generation of classification models from ‘omic’ data. Using pathway analysis methods to better understand the underlying biology of complex diseases such as breast and lung cancers is state-of-the-art. However, the problem of how to combine literature- mining evidence with pathway analysis evidence is an open problem in biomedical informatics research. -
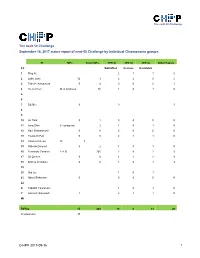
Next-MP50 Status Report
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

ITGB3BP (NM 014288) Human Recombinant Protein – TP300064
OriGene Technologies, Inc. 9620 Medical Center Drive, Ste 200 Rockville, MD 20850, US Phone: +1-888-267-4436 [email protected] EU: [email protected] CN: [email protected] Product datasheet for TP300064 ITGB3BP (NM_014288) Human Recombinant Protein Product data: Product Type: Recombinant Proteins Description: Recombinant protein of human integrin beta 3 binding protein (beta3-endonexin) (ITGB3BP) Species: Human Expression Host: HEK293T Tag: C-Myc/DDK Predicted MW: 20 kDa Concentration: >50 ug/mL as determined by microplate BCA method Purity: > 80% as determined by SDS-PAGE and Coomassie blue staining Buffer: 25 mM Tris.HCl, pH 7.3, 100 mM glycine, 10% glycerol Preparation: Recombinant protein was captured through anti-DDK affinity column followed by conventional chromatography steps. Storage: Store at -80°C. Stability: Stable for 12 months from the date of receipt of the product under proper storage and handling conditions. Avoid repeated freeze-thaw cycles. RefSeq: NP_055103 Locus ID: 23421 UniProt ID: Q13352 RefSeq Size: 1019 Cytogenetics: 1p31.3 RefSeq ORF: 531 Synonyms: CENP-R; CENPR; HSU37139; NRIF3; TAP20 This product is to be used for laboratory only. Not for diagnostic or therapeutic use. View online » ©2021 OriGene Technologies, Inc., 9620 Medical Center Drive, Ste 200, Rockville, MD 20850, US 1 / 2 ITGB3BP (NM_014288) Human Recombinant Protein – TP300064 Summary: This gene encodes a transcriptional coregulator that binds to and enhances the activity of members of the nuclear receptor families, thyroid hormone receptors and retinoid X receptors. This protein also acts as a corepressor of NF-kappaB-dependent signaling. This protein induces apoptosis in breast cancer cells through a caspase 2-mediated signaling pathway. -

Assembly and Analysis of 100 Full MHC Haplotypes from the Danish
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press 1 Assembly and analysis of 100 full MHC haplotypes from the Danish 2 population 3 4 Jacob M. Jensen1*, Palle Villesen1,2, Rune M. Friborg1, The Danish Pan 5 Genome Consortium, Thomas Mailund1, Søren Besenbacher1,3, Mikkel H. 6 Schierup1,4* 7 8 1. Bioinformatics Research Centre, Aarhus University, CF Møllers Alle 8, 9 8000 Aarhus C. Denmark 10 2. Department of Clinical Medicine, Aarhus University, Palle Juul-Jensens 11 Boulevard 82, 8200 Aarhus N. Denmark 12 3. Department of Molecular Medicine, Aarhus University Hospital, Skejby, 13 Aarhus N 8200, Denmark 14 4. Department of Bioscience, Aarhus University, Ny Munkegade 116, 8000 15 Aarhus C. Denmark 16 17 * Correspondence to [email protected], [email protected] 18 19 1 Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press 20 Abstract 21 Genes in the major histocompatibility complex (MHC, also known as 22 HLA) play a critical role in the immune response and variation within the 23 extended 4 Mb region shows association with major risks of many 24 diseases. Yet, deciphering the underlying causes of these associations 25 is difficult because the MHC is the most polymorphic region of the 26 genome with a complex linkage disequilibrium structure. Here we 27 reconstruct full MHC haplotypes from de novo assembled trios without 28 relying on a reference genome and perform evolutionary analyses. We 29 report 100 full MHC haplotypes and call a large set of structural variants 30 in the regions for future use in imputation with GWAS data. -

A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family
Rockefeller University Digital Commons @ RU Student Theses and Dissertations 2018 A High-Throughput Approach to Uncover Novel Roles of APOBEC2, a Functional Orphan of the AID/APOBEC Family Linda Molla Follow this and additional works at: https://digitalcommons.rockefeller.edu/ student_theses_and_dissertations Part of the Life Sciences Commons A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY A Thesis Presented to the Faculty of The Rockefeller University in Partial Fulfillment of the Requirements for the degree of Doctor of Philosophy by Linda Molla June 2018 © Copyright by Linda Molla 2018 A HIGH-THROUGHPUT APPROACH TO UNCOVER NOVEL ROLES OF APOBEC2, A FUNCTIONAL ORPHAN OF THE AID/APOBEC FAMILY Linda Molla, Ph.D. The Rockefeller University 2018 APOBEC2 is a member of the AID/APOBEC cytidine deaminase family of proteins. Unlike most of AID/APOBEC, however, APOBEC2’s function remains elusive. Previous research has implicated APOBEC2 in diverse organisms and cellular processes such as muscle biology (in Mus musculus), regeneration (in Danio rerio), and development (in Xenopus laevis). APOBEC2 has also been implicated in cancer. However the enzymatic activity, substrate or physiological target(s) of APOBEC2 are unknown. For this thesis, I have combined Next Generation Sequencing (NGS) techniques with state-of-the-art molecular biology to determine the physiological targets of APOBEC2. Using a cell culture muscle differentiation system, and RNA sequencing (RNA-Seq) by polyA capture, I demonstrated that unlike the AID/APOBEC family member APOBEC1, APOBEC2 is not an RNA editor. Using the same system combined with enhanced Reduced Representation Bisulfite Sequencing (eRRBS) analyses I showed that, unlike the AID/APOBEC family member AID, APOBEC2 does not act as a 5-methyl-C deaminase. -

The Genetic Program of Pancreatic Beta-Cell Replication in Vivo
Page 1 of 65 Diabetes The genetic program of pancreatic beta-cell replication in vivo Agnes Klochendler1, Inbal Caspi2, Noa Corem1, Maya Moran3, Oriel Friedlich1, Sharona Elgavish4, Yuval Nevo4, Aharon Helman1, Benjamin Glaser5, Amir Eden3, Shalev Itzkovitz2, Yuval Dor1,* 1Department of Developmental Biology and Cancer Research, The Institute for Medical Research Israel-Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 2Department of Molecular Cell Biology, Weizmann Institute of Science, Rehovot, Israel. 3Department of Cell and Developmental Biology, The Silberman Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem 91904, Israel 4Info-CORE, Bioinformatics Unit of the I-CORE Computation Center, The Hebrew University and Hadassah, The Institute for Medical Research Israel- Canada, The Hebrew University-Hadassah Medical School, Jerusalem 91120, Israel 5Endocrinology and Metabolism Service, Department of Internal Medicine, Hadassah-Hebrew University Medical Center, Jerusalem 91120, Israel *Correspondence: [email protected] Running title: The genetic program of pancreatic β-cell replication 1 Diabetes Publish Ahead of Print, published online March 18, 2016 Diabetes Page 2 of 65 Abstract The molecular program underlying infrequent replication of pancreatic beta- cells remains largely inaccessible. Using transgenic mice expressing GFP in cycling cells we sorted live, replicating beta-cells and determined their transcriptome. Replicating beta-cells upregulate hundreds of proliferation- related genes, along with many novel putative cell cycle components. Strikingly, genes involved in beta-cell functions, namely glucose sensing and insulin secretion were repressed. Further studies using single molecule RNA in situ hybridization revealed that in fact, replicating beta-cells double the amount of RNA for most genes, but this upregulation excludes genes involved in beta-cell function. -

Transcriptome Analyses of Lncrnas in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links Between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa
antioxidants Article Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa Luigi Donato 1,2,* , Concetta Scimone 1,2 , Simona Alibrandi 1,3, Carmela Rinaldi 1, Antonina Sidoti 1,2,* and Rosalia D’Angelo 1,2 1 Department of Biomedical and Dental Sciences and Morphofunctional Imaging, Division of Medical Biotechnologies and Preventive Medicine, University of Messina, 98125 Messina, Italy; [email protected] (C.S.); [email protected] (S.A.); [email protected] (C.R.); [email protected] (R.D.) 2 Department of Biomolecular Strategies, Genetics and Avant-Garde Therapies, I.E.ME.S.T., 90139 Palermo, Italy 3 Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, 98125 Messina, Italy * Correspondence: [email protected] (L.D.); [email protected] (A.S.); Tel.: +39-0902213136 (L.D.); +39-0902213372 (A.S.) Received: 21 March 2020; Accepted: 14 April 2020; Published: 15 April 2020 Abstract: Long non-coding RNAs (lncRNAs) are untranslated transcripts which regulate many biological processes. Changes in lncRNA expression pattern are well-known related to various human disorders, such as ocular diseases. Among them, retinitis pigmentosa, one of the most heterogeneous inherited disorder, is strictly related to oxidative stress. However, little is known about regulative aspects able to link oxidative stress to etiopathogenesis of retinitis. Thus, we realized a total RNA-Seq experiment, analyzing human retinal pigment epithelium cells treated by the oxidant agent N-retinylidene-N-retinylethanolamine (A2E), considering three independent experimental groups (untreated control cells, cells treated for 3 h and cells treated for 6 h). -

Computational Simulations to Predict Creatine Kinase-Associated Factors: Protein-Protein Interaction Studies of Brain and Muscle Types of Creatine Kinases
SAGE-Hindawi Access to Research Enzyme Research Volume 2011, Article ID 328249, 12 pages doi:10.4061/2011/328249 Research Article Computational Simulations to Predict Creatine Kinase-Associated Factors: Protein-Protein Interaction Studies of Brain and Muscle Types of Creatine Kinases Wei-Jiang Hu,1 Sheng-Mei Zhou,2 Joshua SungWoo Yang,3, 4 and Fan-Guo Meng1 1 Zhejiang Provincial Key Laboratory of Applied Enzymology, Yangtze Delta Region Institute of Tsinghua University, Jiaxing 314006, China 2 College of Biology and Chemical Engineering, Jiaxing University, Jiaxing 314001, China 3 Korean Bioinformation Center (KOBIC), Korea Research Institute of Bioscience & Biotechnology (KRIBB), Daejeon 305-806, Republic of Korea 4 Department of Bioinformatics, University of Sciences & Technology, Daejeon 205-305, Republic of Korea Correspondence should be addressed to Fan-Guo Meng, [email protected] Received 17 May 2011; Accepted 26 May 2011 Academic Editor: Jun-Mo Yang Copyright © 2011 Wei-Jiang Hu et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. Creatine kinase (CK; EC 2.7.3.2) is related to several skin diseases such as psoriasis and dermatomyositis. CK is important in skin energy homeostasis because it catalyzes the reversible transfer of a phosphoryl group from MgATP to creatine. In this study, we predicted CK binding proteins via the use of bioinformatic tools such as protein-protein interaction (PPI) mappings and suggest the putative hub proteins for CK interactions. We obtained 123 proteins for brain type CK and 85 proteins for muscle type CK in the interaction networks. -

Next-MP50 Stats Report
The neXt-50 Challenge The neXt-50 Challenge Status report and acceptance of next-50 Challenge by individual Chromosome groups April 17, 2017. Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). Status of the Chromosome “parts list”: Step by step milestone plan to find, identify and validate MPs 1. We try to identify more missing proteins through high coverage proteomics technology with testis samples. We will finish this project before May. C-HPP 2017-04-19 1 The neXt-50 Challenge 2. We tested and confirmed that PTM approach could identify significant number of missing proteins. We will finalize the data sets before May. Chromosome 2 See combined response and work plan with Chromosome 14. Chromosome 3 (Takeshi Kawamura) PIC Leaders: Takeshi Kawamura (since January 1) Contributing labs: Major lab members or partners contributing to the neXt50: Takeshi Kawamura: Associate Professor, Proteomics Laboratory, Isotope Science Center, The University of Tokyo, Tokyo, Japan. Toshihide Nishimura: Professor, Department of Translational Medicine Informatics, St. Marianna University School of Medicine, Kanagawa, Japan.