Cnest: a Novel Copy Number Association Discovery Method Uncovers 862 New Associations from 200,629 Whole Exome Sequence Datasets in the UK Biobank
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Extensive Expansion of the Speedy Gene Family in Homininae and Functional Differentiation in Humans
bioRxiv preprint doi: https://doi.org/10.1101/354886; this version posted June 26, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Extensive Expansion of the Speedy gene Family in Homininae and Functional Differentiation in Humans Liang Wang1,2†, Hui Wang1,3,4†, Hongmei Wang1,Yuhui Zhao2, Xiaojun Liu1, Gary Wong5, Qinong Ye6, Xiaoqin Xia7, George F. Gao2, Shan Gao1,8,* 1CAS Key Laboratory of Bio-medical Diagnostics, Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, Suzhou 215163, China; 2CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China; 3 Institute of Biomedical Engineering, Department of Engineering Science, Old Road Campus Research Building, University of Oxford, Oxford OX3 7DQ, UK; 4Oxford Suzhou Centre for Advanced Research (OSCAR), 388 Ruo Shui Road, Suzhou Industrial Park, Jiangsu 215123, China; 5Shenzhen Key Laboratory of Pathogen and Immunity, Guangdong Key Laboratory for Diagnosis and Treatment of Emerging Infectious Diseases, Shenzhen Third People’s Hospital, Shenzhen 518112, China; 6Department of Medical Molecular Biology, Beijing Institute of Biotechnology, Beijing 100850, China; 7Institutes of Hydrobiology, Chinese Academy of Sciences, Wuhan, Hubei, P. R. China, 430072; 8Medical College, Guizhou University, District of Huaxi, Guiyang 550025, China. †These authors contributed equally to this work 1 bioRxiv preprint doi: https://doi.org/10.1101/354886; this version posted June 26, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -
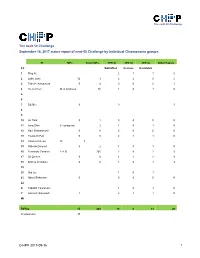
Next-MP50 Status Report
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

Assembly and Analysis of 100 Full MHC Haplotypes from the Danish
Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press 1 Assembly and analysis of 100 full MHC haplotypes from the Danish 2 population 3 4 Jacob M. Jensen1*, Palle Villesen1,2, Rune M. Friborg1, The Danish Pan 5 Genome Consortium, Thomas Mailund1, Søren Besenbacher1,3, Mikkel H. 6 Schierup1,4* 7 8 1. Bioinformatics Research Centre, Aarhus University, CF Møllers Alle 8, 9 8000 Aarhus C. Denmark 10 2. Department of Clinical Medicine, Aarhus University, Palle Juul-Jensens 11 Boulevard 82, 8200 Aarhus N. Denmark 12 3. Department of Molecular Medicine, Aarhus University Hospital, Skejby, 13 Aarhus N 8200, Denmark 14 4. Department of Bioscience, Aarhus University, Ny Munkegade 116, 8000 15 Aarhus C. Denmark 16 17 * Correspondence to [email protected], [email protected] 18 19 1 Downloaded from genome.cshlp.org on October 9, 2021 - Published by Cold Spring Harbor Laboratory Press 20 Abstract 21 Genes in the major histocompatibility complex (MHC, also known as 22 HLA) play a critical role in the immune response and variation within the 23 extended 4 Mb region shows association with major risks of many 24 diseases. Yet, deciphering the underlying causes of these associations 25 is difficult because the MHC is the most polymorphic region of the 26 genome with a complex linkage disequilibrium structure. Here we 27 reconstruct full MHC haplotypes from de novo assembled trios without 28 relying on a reference genome and perform evolutionary analyses. We 29 report 100 full MHC haplotypes and call a large set of structural variants 30 in the regions for future use in imputation with GWAS data. -

Transcriptome Analyses of Lncrnas in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links Between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa
antioxidants Article Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa Luigi Donato 1,2,* , Concetta Scimone 1,2 , Simona Alibrandi 1,3, Carmela Rinaldi 1, Antonina Sidoti 1,2,* and Rosalia D’Angelo 1,2 1 Department of Biomedical and Dental Sciences and Morphofunctional Imaging, Division of Medical Biotechnologies and Preventive Medicine, University of Messina, 98125 Messina, Italy; [email protected] (C.S.); [email protected] (S.A.); [email protected] (C.R.); [email protected] (R.D.) 2 Department of Biomolecular Strategies, Genetics and Avant-Garde Therapies, I.E.ME.S.T., 90139 Palermo, Italy 3 Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, 98125 Messina, Italy * Correspondence: [email protected] (L.D.); [email protected] (A.S.); Tel.: +39-0902213136 (L.D.); +39-0902213372 (A.S.) Received: 21 March 2020; Accepted: 14 April 2020; Published: 15 April 2020 Abstract: Long non-coding RNAs (lncRNAs) are untranslated transcripts which regulate many biological processes. Changes in lncRNA expression pattern are well-known related to various human disorders, such as ocular diseases. Among them, retinitis pigmentosa, one of the most heterogeneous inherited disorder, is strictly related to oxidative stress. However, little is known about regulative aspects able to link oxidative stress to etiopathogenesis of retinitis. Thus, we realized a total RNA-Seq experiment, analyzing human retinal pigment epithelium cells treated by the oxidant agent N-retinylidene-N-retinylethanolamine (A2E), considering three independent experimental groups (untreated control cells, cells treated for 3 h and cells treated for 6 h). -

Development of Novel Analysis and Data Integration Systems to Understand Human Gene Regulation
Development of novel analysis and data integration systems to understand human gene regulation Dissertation zur Erlangung des Doktorgrades Dr. rer. nat. der Fakult¨atf¨urMathematik und Informatik der Georg-August-Universit¨atG¨ottingen im PhD Programme in Computer Science (PCS) der Georg-August University School of Science (GAUSS) vorgelegt von Raza-Ur Rahman aus Pakistan G¨ottingen,April 2018 Prof. Dr. Stefan Bonn, Zentrum f¨urMolekulare Neurobiologie (ZMNH), Betreuungsausschuss: Institut f¨urMedizinische Systembiologie, Hamburg Prof. Dr. Tim Beißbarth, Institut f¨urMedizinische Statistik, Universit¨atsmedizin, Georg-August Universit¨at,G¨ottingen Prof. Dr. Burkhard Morgenstern, Institut f¨urMikrobiologie und Genetik Abtl. Bioinformatik, Georg-August Universit¨at,G¨ottingen Pr¨ufungskommission: Prof. Dr. Stefan Bonn, Zentrum f¨urMolekulare Neurobiologie (ZMNH), Referent: Institut f¨urMedizinische Systembiologie, Hamburg Prof. Dr. Tim Beißbarth, Institut f¨urMedizinische Statistik, Universit¨atsmedizin, Korreferent: Georg-August Universit¨at,G¨ottingen Prof. Dr. Burkhard Morgenstern, Weitere Mitglieder Institut f¨urMikrobiologie und Genetik Abtl. Bioinformatik, der Pr¨ufungskommission: Georg-August Universit¨at,G¨ottingen Prof. Dr. Carsten Damm, Institut f¨urInformatik, Georg-August Universit¨at,G¨ottingen Prof. Dr. Florentin W¨org¨otter, Physikalisches Institut Biophysik, Georg-August-Universit¨at,G¨ottingen Prof. Dr. Stephan Waack, Institut f¨urInformatik, Georg-August Universit¨at,G¨ottingen Tag der m¨undlichen Pr¨ufung: der 30. M¨arz2018 -

Circular DNA Intermediates in the Generation of Large Human Segmental Duplications
Circular DNA intermediates in the generation of large human segmental duplications. Javier Ugarte Chicote IISPV Marcos López-Sánchez Universitat Pompeu Fabra Tomàs Marquès-Bonet Universitat Pompeu Fabra José Callizo Hospital Universitari de Tarragona Joan XXIII Luis Alberto Pérez-Jurado Universitat Pompeu Fabra Antonio Garcia-España ( [email protected] ) iispv https://orcid.org/0000-0002-9957-3161 Research article Keywords: Segmental duplications, circular DNA, human genome evolution, X-Y transposed region, chromoanasynthesis,, MMBIR/FoSTeS, NHEJ, copy number variants Posted Date: July 16th, 2020 DOI: https://doi.org/10.21203/rs.3.rs-27725/v2 License: This work is licensed under a Creative Commons Attribution 4.0 International License. Read Full License Version of Record: A version of this preprint was published on August 26th, 2020. See the published version at https://doi.org/10.1186/s12864-020-06998-w. Page 1/17 Abstract Background: Duplications of large genomic segments provide genetic diversity in genome evolution. Despite their importance, how these duplications are generated remains uncertain, particularly for distant duplicated genomic segments. Results: Here we provide evidence of the participation of circular DNA intermediates in the single generation of some large human segmental duplications. A specic reversion of sequence order from A- B/C-D to B-A/D-C between duplicated segments and the presence of only microhomologies and short indels at the evolutionary breakpoints suggest a circularization of the donor ancestral locus and an accidental replicative interaction with the acceptor locus. Conclusions: This novel mechanism of random genomic mutation could explain several distant genomic duplications including some of the ones that took place during recent human evolution. -

Next-MP50 Stats Report
The neXt-50 Challenge The neXt-50 Challenge Status report and acceptance of next-50 Challenge by individual Chromosome groups April 17, 2017. Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). Status of the Chromosome “parts list”: Step by step milestone plan to find, identify and validate MPs 1. We try to identify more missing proteins through high coverage proteomics technology with testis samples. We will finish this project before May. C-HPP 2017-04-19 1 The neXt-50 Challenge 2. We tested and confirmed that PTM approach could identify significant number of missing proteins. We will finalize the data sets before May. Chromosome 2 See combined response and work plan with Chromosome 14. Chromosome 3 (Takeshi Kawamura) PIC Leaders: Takeshi Kawamura (since January 1) Contributing labs: Major lab members or partners contributing to the neXt50: Takeshi Kawamura: Associate Professor, Proteomics Laboratory, Isotope Science Center, The University of Tokyo, Tokyo, Japan. Toshihide Nishimura: Professor, Department of Translational Medicine Informatics, St. Marianna University School of Medicine, Kanagawa, Japan. -

Supplementary Table 1 Double Treatment Vs Single Treatment
Supplementary table 1 Double treatment vs single treatment Probe ID Symbol Gene name P value Fold change TC0500007292.hg.1 NIM1K NIM1 serine/threonine protein kinase 1.05E-04 5.02 HTA2-neg-47424007_st NA NA 3.44E-03 4.11 HTA2-pos-3475282_st NA NA 3.30E-03 3.24 TC0X00007013.hg.1 MPC1L mitochondrial pyruvate carrier 1-like 5.22E-03 3.21 TC0200010447.hg.1 CASP8 caspase 8, apoptosis-related cysteine peptidase 3.54E-03 2.46 TC0400008390.hg.1 LRIT3 leucine-rich repeat, immunoglobulin-like and transmembrane domains 3 1.86E-03 2.41 TC1700011905.hg.1 DNAH17 dynein, axonemal, heavy chain 17 1.81E-04 2.40 TC0600012064.hg.1 GCM1 glial cells missing homolog 1 (Drosophila) 2.81E-03 2.39 TC0100015789.hg.1 POGZ Transcript Identified by AceView, Entrez Gene ID(s) 23126 3.64E-04 2.38 TC1300010039.hg.1 NEK5 NIMA-related kinase 5 3.39E-03 2.36 TC0900008222.hg.1 STX17 syntaxin 17 1.08E-03 2.29 TC1700012355.hg.1 KRBA2 KRAB-A domain containing 2 5.98E-03 2.28 HTA2-neg-47424044_st NA NA 5.94E-03 2.24 HTA2-neg-47424360_st NA NA 2.12E-03 2.22 TC0800010802.hg.1 C8orf89 chromosome 8 open reading frame 89 6.51E-04 2.20 TC1500010745.hg.1 POLR2M polymerase (RNA) II (DNA directed) polypeptide M 5.19E-03 2.20 TC1500007409.hg.1 GCNT3 glucosaminyl (N-acetyl) transferase 3, mucin type 6.48E-03 2.17 TC2200007132.hg.1 RFPL3 ret finger protein-like 3 5.91E-05 2.17 HTA2-neg-47424024_st NA NA 2.45E-03 2.16 TC0200010474.hg.1 KIAA2012 KIAA2012 5.20E-03 2.16 TC1100007216.hg.1 PRRG4 proline rich Gla (G-carboxyglutamic acid) 4 (transmembrane) 7.43E-03 2.15 TC0400012977.hg.1 SH3D19 -

Genetic Screening Approaches to Cancer Driver Characterization and Synthetic Lethal Target Discovery
Genetic Screening Approaches to Cancer Driver Characterization and Synthetic Lethal Target Discovery The Harvard community has made this article openly available. Please share how this access benefits you. Your story matters Citation Mengwasser, Kristen Elizabeth. 2018. Genetic Screening Approaches to Cancer Driver Characterization and Synthetic Lethal Target Discovery. Doctoral dissertation, Harvard University, Graduate School of Arts & Sciences. Citable link http://nrs.harvard.edu/urn-3:HUL.InstRepos:41121232 Terms of Use This article was downloaded from Harvard University’s DASH repository, and is made available under the terms and conditions applicable to Other Posted Material, as set forth at http:// nrs.harvard.edu/urn-3:HUL.InstRepos:dash.current.terms-of- use#LAA Genetic Screening Approaches to Cancer Driver Characterization and Synthetic Lethal Target Discovery A dissertation presented by Kristen Elizabeth Mengwasser to The Division of Medical Sciences in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the subject of Biological and Biomedical Sciences Harvard University Cambridge, Massachusetts May 2018 © 2018 Kristen Elizabeth Mengwasser All rights reserved. Dissertation Advisor: Dr. Stephen J Elledge Kristen Elizabeth Mengwasser Genetic Screening Approaches to Cancer Driver Characterization and Synthetic Lethal Target Discovery Abstract Advances in genetic screening technology have expanded the toolkit for systematic perturbation of gene function. While the CRISPR-Cas9 system robustly probes genetic loss-of-function in mammalian cells, a barcoded ORFeome library offers the opportunity to systematically study genetic gain-of-function. We employed these two screening tools for three purposes. First, we performed shRNA and CRISPR-based screens for synthetic lethality with BRCA2 deficiency, in two pairs of BRCA2 isogenic cell lines. -

Circular DNA Intermediates in the Generation of Large Human Segmental Duplications
1 Circular DNA intermediates in the generation of large human segmental duplications. 2 Javier U Chicote1, Marcos López-Sánchez 2,3, Tomàs Marquès-Bonet 4,5,6, José Callizo 7, Luis A Pérez- 3 Jurado 2,3,8*, and Antonio García-España1* 4 1 Research Unit, Hospital Universitari de Tarragona Joan XXIII, Institut d’Investigació Sanitària Pere 5 Virgili, Universitat Rovira i Virgili, Tarragona 43005, Spain. 6 2 Genetics Unit, Departament de Ciències Experimentals i de la Salut, Universitat Pompeu Fabra, 7 Barcelona 08003, Spain. 8 3 Hospital del Mar Research Institute (IMIM) and Centro de Investigación Biomédica en Red de 9 Enfermedades Raras (CIBERER), Barcelona 08003, Spain. 10 4 Institut de Biologia Evolutiva (CSIC-UPF), Departament de Ciències Experimentals i de la Salut, 11 Universitat Pompeu Fabra, Barcelona 08003, Spain. 12 5 Catalan Institution of Research and Advanced Studies (ICREA), Barcelona 08010, Spain 13 6 CNAG-CRG, Centre for Genomic Regulation, Barcelona Institute of Science and Technology (BIST), 14 Barcelona 08028, Spain. 15 7 Department of Ophthalmology, Hospital Universitari de Tarragona Joan XXIII, Institut d’Investigació 16 Sanitària Pere Virgili, Universitat Rovira i Virgili, Tarragona 43005, Spain. 17 8 SA Clinical Genetics, Women's and Children's Hospital, South Australian Health and Medical Research 18 Institute (SAHMRI) & University of Adelaide, Adelaide, SA 5000, Australia. 19 20 *Corresponding author: [email protected] (A.G-E); [email protected] (L.A.P-J) 21 22 RUNNING TITLE: Human genomic duplications by circular DNA 23 24 25 Keywords 26 Segmental duplications, circular DNA, human genome evolution, X-Y transposed region, 27 chromoanasynthesis,, MMBIR/FoSTeS, NHEJ, copy number variants. -

MECHANISMS of TUMORIGENESIS in AFRICAN AMERICAN COLORECTAL CANCER by Gaius J. Augustus
Mechanisms of Tumorigenesis in African American Colorectal Cancer Item Type text; Electronic Dissertation Authors Augustus, Gaius Julian Publisher The University of Arizona. Rights Copyright © is held by the author. Digital access to this material is made possible by the University Libraries, University of Arizona. Further transmission, reproduction, presentation (such as public display or performance) of protected items is prohibited except with permission of the author. Download date 27/09/2021 11:20:21 Link to Item http://hdl.handle.net/10150/633006 MECHANISMS OF TUMORIGENESIS IN AFRICAN AMERICAN COLORECTAL CANCER by Gaius J. Augustus __________________________ Copyright © Gaius J. Augustus 2019 A Dissertation Submitted to the Faculty of the GRADUATE INTERDISCIPLINARY PROGRAM IN CANCER BIOLOGY In Partial Fulfillment of the Requirements For the Degree of DOCTOR OF PHILOSOPHY In the Graduate College THE UNIVERSITY OF ARIZONA 2019 Mechanisms of Tumorigenesis in African American CRC 2 Mechanisms of Tumorigenesis in African American CRC Acknowledgements This work was supported by grants from the National Cancer Institute (U01 CA153060 and P30 CA023074, NAE; RO1 CA204808, HRG, EM, LTH; RO1 CA141057, BJ) and the American Cancer Society Illinois Division (223187, XL). GJA was supported by a Cancer Biology Training Grant (T32CA009213). The funders had no role in the design of the study; the collection, analysis, or interpretation of the data; the writing of the manuscript; or the decision to submit the manuscript for publication. The author gratefully acknowledges the recruiters of the CCCC for their dedication and integrity, including Maggie Moran, Timothy Carroll, Katy Ceryes, Amy Disharoon, Archana Krishnan, Katie Morrissey, Maureen Regan, and Katya Seligman.