Neurophysiological and Genetic Findings in Patients with Juvenile Myoclonic Epilepsy
Total Page:16
File Type:pdf, Size:1020Kb

Load more
Recommended publications
-

Contiguous Deletion of the NDP, MAOA, MAOB, and EFHC2 Genes in a Patient with Norrie Disease, Severe Psychomotor Retardation and Myoclonic Epilepsy
ß 2007 Wiley-Liss, Inc. American Journal of Medical Genetics Part A 143A:916–920 (2007) Contiguous Deletion of the NDP, MAOA, MAOB, and EFHC2 Genes in a Patient With Norrie Disease, Severe Psychomotor Retardation and Myoclonic Epilepsy L. Rodriguez-Revenga,1,2 I. Madrigal,1,2 L.S. Alkhalidi,3 L. Armengol,4 E. Gonza´lez,4 C. Badenas,1,2 X. Estivill,1,5 and M. Mila`1,2* 1Biochemistry and Molecular Genetics Department, Hospital Clı´nic, Barcelona, Spain 2IDIBAPS (Institut d’Investigacions Biome`diques August Pi i Sunyer), Barcelona, Spain 3Department of Health and Medical Services, Rashid Hospital, Dubai, United Arab Emirates 4Genes and Disease Programme, Centre for Genomic Regulation (CRG), Barcelona Biomedical Research Park, Barcelona, Spain 5Department of Experimental and Health Sciences, Universitat Pompeu Fabra (UPF), Barcelona, Spain Received 20 February 2006; Accepted 7 September 2006 Norrie disease (ND) is an X-linked disorder, inherited as a Clinical features of the proband include bilateral retinal recessive trait that, therefore, mostly affects males. The gene detachment, microcephaly, severe psychomotor retardation responsible for ND, called NDP, maps to the short arm of without verbal language skills acquired, and epilepsy. The chromosome X (Xp11.4-p11.3). We report here an atypical identification and molecular characterization of this case case of ND, consisting of a patient harboring a large reinforces the idea of a new contiguous gene syndrome that submicroscopic deletion affecting not only the NDP gene would explain the complex phenotype shared by atypical but also the MAOA, MAOB, and EFHC2 genes. Microarray ND patients. ß 2007 Wiley-Liss, Inc. -

Clinicians Using the Classification Will Identify a Seizure As Focal Or Generalized Onset If There Is About an 80% Confidence Level About the Type of Onset
GENERALIZED ONSET SEIZURES Generalized onset seizures are not characterized by level of awareness, because awareness is almost always impaired. Generalized tonic-clonic: Immediate loss of Generalized epileptic spasms: Brief seizures with awareness, with stiffening of all limbs (tonic phase), flexion at the trunk and flexion or extension of the followed by sustained rhythmic jerking of limbs and limbs. Video-EEG recording may be required to face (clonic phase). Duration is typically 1 to 3 minutes. determine focal versus generalized onset. The seizure may produce a cry at the start, falling, tongue biting, and incontinence. Generalized typical absence: Sudden onset when activity stops with a brief pause and staring, Generalized clonic: Rhythmical sustained jerking of sometimes with eye fluttering and head nodding or limbs and/or head with no tonic stiffening phase. other automatic behaviors. If it lasts for more than These seizures most often occur in young children. several seconds, awareness and memory are impaired. Recovery is immediate. The EEG during these seizures Generalized tonic: Stiffening of all limbs, without always shows generalized spike-waves. clonic jerking. Generalized atypical absence: Like typical absence Generalized myoclonic: Irregular, unsustained jerking seizures, but may have slower onset and recovery and of limbs, face, eyes, or eyelids. The jerking of more pronounced changes in tone. Atypical absence generalized myoclonus may not always be left-right seizures can be difficult to distinguish from focal synchronous, but it occurs on both sides. impaired awareness seizures, but absence seizures usually recover more quickly and the EEG patterns are Generalized myoclonic-tonic-clonic: This seizure is like different. -
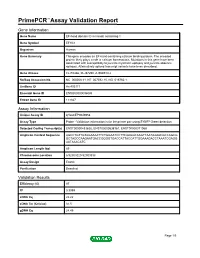
Primepcr™Assay Validation Report
PrimePCR™Assay Validation Report Gene Information Gene Name EF-hand domain (C-terminal) containing 1 Gene Symbol EFHC1 Organism Human Gene Summary This gene encodes an EF-hand-containing calcium binding protein. The encoded protein likely plays a role in calcium homeostasis. Mutations in this gene have been associated with susceptibility to juvenile myoclonic epilepsy and juvenile absence epilepsy. Alternatively spliced transcript variants have been described. Gene Aliases FLJ10466, FLJ37290, dJ304B14.2 RefSeq Accession No. NC_000006.11, NT_007592.15, NG_016760.1 UniGene ID Hs.403171 Ensembl Gene ID ENSG00000096093 Entrez Gene ID 114327 Assay Information Unique Assay ID qHsaCEP0049958 Assay Type Probe - Validation information is for the primer pair using SYBR® Green detection Detected Coding Transcript(s) ENST00000433625, ENST00000538167, ENST00000371068 Amplicon Context Sequence AGCCTGTTGTAGAAAATTCTGGAATCCTTCAAGGCAAGTTAATAAAACGCCAGCG GCTAGCCAAGAATGACCGGGGTGACCATTACCATTGGAAAGACCTAAATCGAGG AATAAACATC Amplicon Length (bp) 89 Chromosome Location 6:52303220-52303338 Assay Design Exonic Purification Desalted Validation Results Efficiency (%) 97 R2 0.9999 cDNA Cq 22.22 cDNA Tm (Celsius) 81.5 gDNA Cq 24.45 Page 1/5 PrimePCR™Assay Validation Report Specificity (%) 100 Information to assist with data interpretation is provided at the end of this report. Page 2/5 PrimePCR™Assay Validation Report EFHC1, Human Amplification Plot Amplification of cDNA generated from 25 ng of universal reference RNA Melt Peak Melt curve analysis of above amplification -

Neuropathology Category Code List
Neuropathology Page 1 of 27 Neuropathology Major Category Code Headings Revised 10/2018 1 General neuroanatomy, pathology, and staining 65000 2 Developmental neuropathology, NOS 65400 3 Epilepsy 66230 4 Vascular disorders 66300 5 Trauma 66600 6 Infectious/inflammatory disease 66750 7 Demyelinating diseases 67200 8 Complications of systemic disorders 67300 9 Aging and neurodegenerative diseases 68000 10 Prion diseases 68400 11 Neoplasms 68500 12 Skeletal Muscle 69500 13 Peripheral Nerve 69800 14 Ophthalmic pathology 69910 Neuropathology Page 2 of 27 Neuropathology 1 General neuroanatomy, pathology, and staining 65000 A Neuroanatomy, NOS 65010 1 Neocortex 65011 2 White matter 65012 3 Entorhinal cortex/hippocampus 65013 4 Deep (basal) nuclei 65014 5 Brain stem 65015 6 Cerebellum 65016 7 Spinal cord 65017 8 Pituitary 65018 9 Pineal 65019 10 Tracts 65020 11 Vascular supply 65021 12 Notochord 65022 B Cell types 65030 1 Neurons 65031 2 Astrocytes 65032 3 Oligodendroglia 65033 4 Ependyma 65034 5 Microglia and mononuclear cells 65035 6 Choroid plexus 65036 7 Meninges 65037 8 Blood vessels 65038 C Cerebrospinal fluid 65045 D Pathologic responses in neurons and axons 65050 1 Axonal degeneration/spheroid/reaction 65051 2 Central chromatolysis 65052 3 Tract degeneration 65053 4 Swollen/ballooned neurons 65054 5 Trans-synaptic neuronal degeneration 65055 6 Olivary hypertrophy 65056 7 Acute ischemic (hypoxic) cell change 65057 8 Apoptosis 65058 9 Protein aggregation 65059 10 Protein degradation/ubiquitin pathway 65060 E Neuronal nuclear inclusions 65100 -

Myoclonic Status Epilepticus in Juvenile Myoclonic Epilepsy
Original article Epileptic Disord 2009; 11 (4): 309-14 Myoclonic status epilepticus in juvenile myoclonic epilepsy Julia Larch, Iris Unterberger, Gerhard Bauer, Johannes Reichsoellner, Giorgi Kuchukhidze, Eugen Trinka Department of Neurology, Medical University of Innsbruck, Austria Received April 9, 2009; Accepted November 18, 2009 ABSTRACT – Background. Myoclonic status epilepticus (MSE) is rarely found in juvenile myoclonic epilepsy (JME) and its clinical features are not well described. We aimed to analyze MSE incidence, precipitating factors and clini- cal course by studying patients with JME from a large outpatient epilepsy clinic. Methods. We retrospectively screened all patients with JME treated at the Department of Neurology, Medical University of Innsbruck, Austria between 1970 and 2007 for a history of MSE. We analyzed age, sex, age at seizure onset, seizure types, EEG, MRI/CT findings and response to antiepileptic drugs. Results. Seven patients (five women, two men; median age at time of MSE 31 years; range 17-73) with MSE out of a total of 247 patients with JME were identi- fied. The median follow-up time was seven years (range 0-35), the incidence was 3.2/1,000 patient years. Median duration of epilepsy before MSE was 26 years (range 10-58). We identified three subtypes: 1) MSE with myoclonic seizures only in two patients, 2) MSE with generalized tonic clonic seizures in three, and 3) generalized tonic clonic seizures with myoclonic absence status in two patients. All patients responded promptly to benzodiazepines. One patient had repeated episodes of MSE. Precipitating events were identified in all but one patient. Drug withdrawal was identified in four patients, one of whom had additional sleep deprivation and alcohol intake. -

Supplementary Table S4. FGA Co-Expressed Gene List in LUAD
Supplementary Table S4. FGA co-expressed gene list in LUAD tumors Symbol R Locus Description FGG 0.919 4q28 fibrinogen gamma chain FGL1 0.635 8p22 fibrinogen-like 1 SLC7A2 0.536 8p22 solute carrier family 7 (cationic amino acid transporter, y+ system), member 2 DUSP4 0.521 8p12-p11 dual specificity phosphatase 4 HAL 0.51 12q22-q24.1histidine ammonia-lyase PDE4D 0.499 5q12 phosphodiesterase 4D, cAMP-specific FURIN 0.497 15q26.1 furin (paired basic amino acid cleaving enzyme) CPS1 0.49 2q35 carbamoyl-phosphate synthase 1, mitochondrial TESC 0.478 12q24.22 tescalcin INHA 0.465 2q35 inhibin, alpha S100P 0.461 4p16 S100 calcium binding protein P VPS37A 0.447 8p22 vacuolar protein sorting 37 homolog A (S. cerevisiae) SLC16A14 0.447 2q36.3 solute carrier family 16, member 14 PPARGC1A 0.443 4p15.1 peroxisome proliferator-activated receptor gamma, coactivator 1 alpha SIK1 0.435 21q22.3 salt-inducible kinase 1 IRS2 0.434 13q34 insulin receptor substrate 2 RND1 0.433 12q12 Rho family GTPase 1 HGD 0.433 3q13.33 homogentisate 1,2-dioxygenase PTP4A1 0.432 6q12 protein tyrosine phosphatase type IVA, member 1 C8orf4 0.428 8p11.2 chromosome 8 open reading frame 4 DDC 0.427 7p12.2 dopa decarboxylase (aromatic L-amino acid decarboxylase) TACC2 0.427 10q26 transforming, acidic coiled-coil containing protein 2 MUC13 0.422 3q21.2 mucin 13, cell surface associated C5 0.412 9q33-q34 complement component 5 NR4A2 0.412 2q22-q23 nuclear receptor subfamily 4, group A, member 2 EYS 0.411 6q12 eyes shut homolog (Drosophila) GPX2 0.406 14q24.1 glutathione peroxidase -

Neuronal Hyperexcitability in a Mouse Model of SCN8A Epileptic Encephalopathy
Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy Luis F. Lopez-Santiagoa,1, Yukun Yuana,1, Jacy L. Wagnonb, Jacob M. Hulla, Chad R. Frasiera, Heather A. O’Malleya, Miriam H. Meislerb,c, and Lori L. Isoma,c,d,2 aDepartment of Pharmacology, University of Michigan, Ann Arbor, MI 48109; bDepartment of Human Genetics, University of Michigan, Ann Arbor, MI 48109; cDepartment of Neurology, University of Michigan, Ann Arbor, MI 48109; and dDepartment of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI 48109 Edited by William A. Catterall, University of Washington School of Medicine, Seattle, WA, and approved January 17, 2017 (received for review October 12, 2016) Patients with early infantile epileptic encephalopathy (EIEE) experi- include seizure onset between birth and 18 mo of age, mild to se- ence severe seizures and cognitive impairment and are at increased vere cognitive and developmental delay, and mild to severe risk for sudden unexpected death in epilepsy (SUDEP). EIEE13 [Online movement disorders (11, 13). Approximately 50% of patients are Mendelian Inheritance in Man (OMIM) # 614558] is caused by de nonambulatory and 12% of published cases (5/43) experienced novo missense mutations in the voltage-gated sodium channel gene SUDEP during childhood or adolescence (10, 13, 16, 17). SCN8A. Here, we investigated the neuronal phenotype of a mouse Almost all of the identified mutations in SCN8A are missense model expressing the gain-of-function SCN8A patient mutation, mutations. Ten of these mutations have been tested functionally in p.Asn1768Asp (Nav1.6-N1768D). Our results revealed regional transfected cells, and eight were found to introduce changes in the and neuronal subtype specificity in the effects of the N1768D muta- biophysical properties of Na 1.6 that are predicted to result in Scn8aN1768D/+ v tion. -

Whole Exome Sequencing in Families at High Risk for Hodgkin Lymphoma: Identification of a Predisposing Mutation in the KDR Gene
Hodgkin Lymphoma SUPPLEMENTARY APPENDIX Whole exome sequencing in families at high risk for Hodgkin lymphoma: identification of a predisposing mutation in the KDR gene Melissa Rotunno, 1 Mary L. McMaster, 1 Joseph Boland, 2 Sara Bass, 2 Xijun Zhang, 2 Laurie Burdett, 2 Belynda Hicks, 2 Sarangan Ravichandran, 3 Brian T. Luke, 3 Meredith Yeager, 2 Laura Fontaine, 4 Paula L. Hyland, 1 Alisa M. Goldstein, 1 NCI DCEG Cancer Sequencing Working Group, NCI DCEG Cancer Genomics Research Laboratory, Stephen J. Chanock, 5 Neil E. Caporaso, 1 Margaret A. Tucker, 6 and Lynn R. Goldin 1 1Genetic Epidemiology Branch, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 2Cancer Genomics Research Laboratory, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; 3Ad - vanced Biomedical Computing Center, Leidos Biomedical Research Inc.; Frederick National Laboratory for Cancer Research, Frederick, MD; 4Westat, Inc., Rockville MD; 5Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD; and 6Human Genetics Program, Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH, Bethesda, MD, USA ©2016 Ferrata Storti Foundation. This is an open-access paper. doi:10.3324/haematol.2015.135475 Received: August 19, 2015. Accepted: January 7, 2016. Pre-published: June 13, 2016. Correspondence: [email protected] Supplemental Author Information: NCI DCEG Cancer Sequencing Working Group: Mark H. Greene, Allan Hildesheim, Nan Hu, Maria Theresa Landi, Jennifer Loud, Phuong Mai, Lisa Mirabello, Lindsay Morton, Dilys Parry, Anand Pathak, Douglas R. Stewart, Philip R. Taylor, Geoffrey S. Tobias, Xiaohong R. Yang, Guoqin Yu NCI DCEG Cancer Genomics Research Laboratory: Salma Chowdhury, Michael Cullen, Casey Dagnall, Herbert Higson, Amy A. -

Extensive Expansion of the Speedy Gene Family in Homininae and Functional Differentiation in Humans
bioRxiv preprint doi: https://doi.org/10.1101/354886; this version posted June 26, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY-NC-ND 4.0 International license. Extensive Expansion of the Speedy gene Family in Homininae and Functional Differentiation in Humans Liang Wang1,2†, Hui Wang1,3,4†, Hongmei Wang1,Yuhui Zhao2, Xiaojun Liu1, Gary Wong5, Qinong Ye6, Xiaoqin Xia7, George F. Gao2, Shan Gao1,8,* 1CAS Key Laboratory of Bio-medical Diagnostics, Suzhou Institute of Biomedical Engineering and Technology, Chinese Academy of Sciences, Suzhou 215163, China; 2CAS Key Laboratory of Pathogenic Microbiology and Immunology, Institute of Microbiology, Chinese Academy of Sciences, Beijing 100101, China; 3 Institute of Biomedical Engineering, Department of Engineering Science, Old Road Campus Research Building, University of Oxford, Oxford OX3 7DQ, UK; 4Oxford Suzhou Centre for Advanced Research (OSCAR), 388 Ruo Shui Road, Suzhou Industrial Park, Jiangsu 215123, China; 5Shenzhen Key Laboratory of Pathogen and Immunity, Guangdong Key Laboratory for Diagnosis and Treatment of Emerging Infectious Diseases, Shenzhen Third People’s Hospital, Shenzhen 518112, China; 6Department of Medical Molecular Biology, Beijing Institute of Biotechnology, Beijing 100850, China; 7Institutes of Hydrobiology, Chinese Academy of Sciences, Wuhan, Hubei, P. R. China, 430072; 8Medical College, Guizhou University, District of Huaxi, Guiyang 550025, China. †These authors contributed equally to this work 1 bioRxiv preprint doi: https://doi.org/10.1101/354886; this version posted June 26, 2018. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. -

Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications
Stem Cell Rev and Rep DOI 10.1007/s12015-016-9662-8 Detailed Characterization of Human Induced Pluripotent Stem Cells Manufactured for Therapeutic Applications Behnam Ahmadian Baghbaderani 1 & Adhikarla Syama2 & Renuka Sivapatham3 & Ying Pei4 & Odity Mukherjee2 & Thomas Fellner1 & Xianmin Zeng3,4 & Mahendra S. Rao5,6 # The Author(s) 2016. This article is published with open access at Springerlink.com Abstract We have recently described manufacturing of hu- help determine which set of tests will be most useful in mon- man induced pluripotent stem cells (iPSC) master cell banks itoring the cells and establishing criteria for discarding a line. (MCB) generated by a clinically compliant process using cord blood as a starting material (Baghbaderani et al. in Stem Cell Keywords Induced pluripotent stem cells . Embryonic stem Reports, 5(4), 647–659, 2015). In this manuscript, we de- cells . Manufacturing . cGMP . Consent . Markers scribe the detailed characterization of the two iPSC clones generated using this process, including whole genome se- quencing (WGS), microarray, and comparative genomic hy- Introduction bridization (aCGH) single nucleotide polymorphism (SNP) analysis. We compare their profiles with a proposed calibra- Induced pluripotent stem cells (iPSCs) are akin to embryonic tion material and with a reporter subclone and lines made by a stem cells (ESC) [2] in their developmental potential, but dif- similar process from different donors. We believe that iPSCs fer from ESC in the starting cell used and the requirement of a are likely to be used to make multiple clinical products. We set of proteins to induce pluripotency [3]. Although function- further believe that the lines used as input material will be used ally identical, iPSCs may differ from ESC in subtle ways, at different sites and, given their immortal status, will be used including in their epigenetic profile, exposure to the environ- for many years or even decades. -

Reviews Familial Cortical Myoclonic Tremor and Epilepsy, an Enigmatic Disorder: from Phenotypes to Pathophysiology and Genetics
Freely available online Reviews Familial Cortical Myoclonic Tremor and Epilepsy, an Enigmatic Disorder: From Phenotypes to Pathophysiology and Genetics. A Systematic Review 1 1 2,3 1* Tom van den Ende , Sarvi Sharifi , Sandra M. A. van der Salm & Anne-Fleur van Rootselaar 1 Department of Neurology and Clinical Neurophysiology, Amsterdam Neuroscience, Academic Medical Center, Amsterdam, The Netherlands, 2 Brain Center Rudolf Magnus, Department of Neurology and Neurosurgery, University Medical Center, Utrecht, The Netherlands, 3 Stichting Epilepsie Instellingen Nederland (SEIN), Zwolle, The Netherlands Abstract Background: Autosomal dominant familial cortical myoclonic tremor and epilepsy (FCMTE) is characterized by distal tremulous myoclonus, generalized seizures, and signs of cortical reflex myoclonus. FCMTE has been described in over 100 pedigrees worldwide, under several different names and acronyms. Pathological changes have been located in the cerebellum. This systematic review discusses the clinical spectrum, treatment, pathophysiology, and genetic findings. Methods: We carried out a PubMed search, using a combination of the following search terms: cortical tremor, myoclonus, epilepsy, benign course, adult onset, familial, and autosomal dominant; this resulted in a total of 77 studies (761 patients; 126 pedigrees) fulfilling the inclusion and exclusion criteria. Results: Phenotypic differences across pedigrees exist, possibly related to underlying genetic differences. A ‘‘benign’’ phenotype has been described in several Japanese families and pedigrees linked to 8q (FCMTE1). French patients (5p linkage; FCMTE3) exhibit more severe progression, and in Japanese/Chinese pedigrees (with unknown linkage) anticipation has been suggested. Preferred treatment is with valproate (mind teratogenicity), levetiracetam, and/or clonazepam. Several genes have been identified, which differ in potential pathogenicity. Discussion: Based on the core features (above), the syndrome can be considered a distinct clinical entity. -

Novel Gene Discovery in Primary Ciliary Dyskinesia
Novel Gene Discovery in Primary Ciliary Dyskinesia Mahmoud Raafat Fassad Genetics and Genomic Medicine Programme Great Ormond Street Institute of Child Health University College London A thesis submitted in conformity with the requirements for the degree of Doctor of Philosophy University College London 1 Declaration I, Mahmoud Raafat Fassad, confirm that the work presented in this thesis is my own. Where information has been derived from other sources, I confirm that this has been indicated in the thesis. 2 Abstract Primary Ciliary Dyskinesia (PCD) is one of the ‘ciliopathies’, genetic disorders affecting either cilia structure or function. PCD is a rare recessive disease caused by defective motile cilia. Affected individuals manifest with neonatal respiratory distress, chronic wet cough, upper respiratory tract problems, progressive lung disease resulting in bronchiectasis, laterality problems including heart defects and adult infertility. Early diagnosis and management are essential for better respiratory disease prognosis. PCD is a highly genetically heterogeneous disorder with causal mutations identified in 36 genes that account for the disease in about 70% of PCD cases, suggesting that additional genes remain to be discovered. Targeted next generation sequencing was used for genetic screening of a cohort of patients with confirmed or suggestive PCD diagnosis. The use of multi-gene panel sequencing yielded a high diagnostic output (> 70%) with mutations identified in known PCD genes. Over half of these mutations were novel alleles, expanding the mutation spectrum in PCD genes. The inclusion of patients from various ethnic backgrounds revealed a striking impact of ethnicity on the composition of disease alleles uncovering a significant genetic stratification of PCD in different populations.