Assembly and Analysis of 100 Full MHC Haplotypes from the Danish
Total Page:16
File Type:pdf, Size:1020Kb
Load more
Recommended publications
-
Next-MP50 Status Report
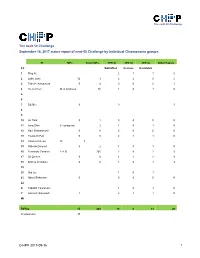
The neXt-50 Challenge The neXt-50 Challenge September 16, 2017 status report of next-50 Challenge by individual Chromosome groups. PI MPs Silver MPs JPR SI JPR SI JPR SI Other Papers Ch Submitted In press In revision 1 Ping Xu 2 1 1 0 2 Lydie Lane 12 3 2 2 0 2 3 Takeshi Kawamura 0 4 0 0 0 1 4 Yu-Ju Chen 26 in progress 86 1 0 1 0 5 6 7 Ed Nice 0 0 1 8 9 10 Jin Park 0 1 0 0 0 0 11 Jong Shin 2 in progress 2 1 0 1 0 12 Ravi Sirdeshmukh 0 0 0 0 0 0 13 Young-Ki Paik 0 5 2 1 1 0 14 CharLes Pineau 12 3 15 GiLberto Domont 0 2 1 0 1 0 16 Fernando CorraLes 2 (+ 9) 165 2 0 2 5 17 GiL Omenn 0 0 3 1 2 8 18 Andrey Archakov 0 0 1 0 1 3 19 20 Siqi Liu 1 0 1 21 Albert Sickmann 0 0 0 0 0 22 X Tadashi Yamamoto 1 0 1 0 Y Hosseini SaLekdeh 1 2 1 1 0 Mt TOTAL 15 268 19 6 13 20 (in progress) 37 C-HPP 2017-09-16 1 The neXt-50 Challenge Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). -

Transcriptome Analyses of Lncrnas in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links Between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa
antioxidants Article Transcriptome Analyses of lncRNAs in A2E-Stressed Retinal Epithelial Cells Unveil Advanced Links between Metabolic Impairments Related to Oxidative Stress and Retinitis Pigmentosa Luigi Donato 1,2,* , Concetta Scimone 1,2 , Simona Alibrandi 1,3, Carmela Rinaldi 1, Antonina Sidoti 1,2,* and Rosalia D’Angelo 1,2 1 Department of Biomedical and Dental Sciences and Morphofunctional Imaging, Division of Medical Biotechnologies and Preventive Medicine, University of Messina, 98125 Messina, Italy; [email protected] (C.S.); [email protected] (S.A.); [email protected] (C.R.); [email protected] (R.D.) 2 Department of Biomolecular Strategies, Genetics and Avant-Garde Therapies, I.E.ME.S.T., 90139 Palermo, Italy 3 Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, University of Messina, 98125 Messina, Italy * Correspondence: [email protected] (L.D.); [email protected] (A.S.); Tel.: +39-0902213136 (L.D.); +39-0902213372 (A.S.) Received: 21 March 2020; Accepted: 14 April 2020; Published: 15 April 2020 Abstract: Long non-coding RNAs (lncRNAs) are untranslated transcripts which regulate many biological processes. Changes in lncRNA expression pattern are well-known related to various human disorders, such as ocular diseases. Among them, retinitis pigmentosa, one of the most heterogeneous inherited disorder, is strictly related to oxidative stress. However, little is known about regulative aspects able to link oxidative stress to etiopathogenesis of retinitis. Thus, we realized a total RNA-Seq experiment, analyzing human retinal pigment epithelium cells treated by the oxidant agent N-retinylidene-N-retinylethanolamine (A2E), considering three independent experimental groups (untreated control cells, cells treated for 3 h and cells treated for 6 h). -

Next-MP50 Stats Report
The neXt-50 Challenge The neXt-50 Challenge Status report and acceptance of next-50 Challenge by individual Chromosome groups April 17, 2017. Chromosome 1 (Ping Xu) PIC Leaders: Ping Xu, Fuchu He Contributing labs: Ping Xu, Beijing Proteome Research Center Fuchu He, Beijing Proteome Research Center Dong Yang, Beijing Proteome Research Center Wantao Ying, Beijing Proteome Research Center Pengyuan Yang, Fudan University Siqi Liu, Beijing Genome Institute Qinyu He, Jinan University Major lab members or partners contributing to the neXt50: Yao Zhang (Beijing Proteome Research Center), Yihao Wang (Beijing Proteome Research Center), Cuitong He (Beijing Proteome Research Center), Wei Wei (Beijing Proteome Research Center), Yanchang Li (Beijing Proteome Research Center), Feng Xu (Beijing Proteome Research Center), Xuehui Peng (Beijing Proteome Research Center). Status of the Chromosome “parts list”: Step by step milestone plan to find, identify and validate MPs 1. We try to identify more missing proteins through high coverage proteomics technology with testis samples. We will finish this project before May. C-HPP 2017-04-19 1 The neXt-50 Challenge 2. We tested and confirmed that PTM approach could identify significant number of missing proteins. We will finalize the data sets before May. Chromosome 2 See combined response and work plan with Chromosome 14. Chromosome 3 (Takeshi Kawamura) PIC Leaders: Takeshi Kawamura (since January 1) Contributing labs: Major lab members or partners contributing to the neXt50: Takeshi Kawamura: Associate Professor, Proteomics Laboratory, Isotope Science Center, The University of Tokyo, Tokyo, Japan. Toshihide Nishimura: Professor, Department of Translational Medicine Informatics, St. Marianna University School of Medicine, Kanagawa, Japan. -

1 SUPPLEMENTARY METHODS Scoring the Schizophrenia Risk Gene
SUPPLEMENTARY METHODS Scoring the schizophrenia risk gene candidates We have developed a statistical method to score the disease-relatedness of candidate genes with predictive features extracted from gene networks and annotation based on a set of training disease genes using frequent item set mining algorithm (Figure S1). For schizophrenia, we will first curate a set of genes, D, known to be associated with this disease from the SZGR database (JIA et al. 2010). Given D and the set of all known genes G (from GENCODE v19), we obtain the background genes B = G – D. First, from D we will extract the predictive features – i.e., the frequent combinations of either the direct neighbors of schizophrenia genes in the functional linkage network (LINGHU et al. 2009) (with the functional linkage weight cutoff = 1) or the gene ontology (GO) terms of schizophrenia genes – using the frequent item set mining algorithm (AGRAWAL et al. 1995) (with the support = 0.093) . GO terms of schizophrenia genes include not only annotated GO terms but also their ancestors GO terms along the paths of the “is a” relationship in the GO hierarchy structure. The considered predictive features are limit to frequent combinations with sizes no greater than 3 to avoid redundancy and intensive computation. Then, each predictive feature will be scored by the frequency with which it appears in D and B: 푆푓 = (퐹퐷⁄푁퐷)⁄(퐹퐵⁄푁퐵) (1), in which FD is the frequency with which the predictive feature, f, occurs in D and ND the number of genes in D. FB and NB have similar meanings. -

Cnest: a Novel Copy Number Association Discovery Method Uncovers 862 New Associations from 200,629 Whole Exome Sequence Datasets in the UK Biobank
bioRxiv preprint doi: https://doi.org/10.1101/2021.08.19.456963; this version posted August 19, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. CNest: A Novel Copy Number Association Discovery Method Uncovers 862 New Associations from 200,629 Whole Exome Sequence Datasets in the UK Biobank Tomas Fitzgerald1* & Ewan Birney1* 1) European Molecular Biology Laboratory, European Bioinformatics Institute (EMBL- EBI), Wellcome Genome Campus, Cambridge, CB10 1SD, UK * Corresponding authors: Tomas William Fitzgerald, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Cambridge, CB10 1SD, UK, Tel:, Fax:, Email: [email protected] Ewan Birney, European Bioinformatics Institute (EMBL-EBI), Wellcome Genome Campus, Cambridge, CB10 1SD, UK, Tel:, Fax:, Email: [email protected] bioRxiv preprint doi: https://doi.org/10.1101/2021.08.19.456963; this version posted August 19, 2021. The copyright holder for this preprint (which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made available under aCC-BY 4.0 International license. Abstract Copy number variation (CNV) has long been known to influence human traits having a rich history of research into common and rare genetic disease and although CNV is accepted as an important class of genomic variation, progress on copy number (CN) phenotype associations from Next Generation Sequencing data (NGS) has been limited, in part, due to the relative difficulty in CNV detection and an enrichment for large numbers of false positives. -

The Role of Epigenetic Modifications, Long-Range Contacts, Enhancers
www.nature.com/scientificreports OPEN The role of epigenetic modifcations, long‑range contacts, enhancers and topologically associating domains in the regulation of glioma grade‑specifc genes Ilona E. Grabowicz1*, Bartek Wilczyński2, Bożena Kamińska3, Adria‑Jaume Roura3, Bartosz Wojtaś3 & Michał J. Dąbrowski1* Genome‑wide studies have uncovered specifc genetic alterations, transcriptomic patterns and epigenetic profles associated with diferent glioma types. We have recently created a unique atlas encompassing genome‑wide profles of open chromatin, histone H3K27ac and H3Kme3 modifcations, DNA methylation and transcriptomes of 33 glioma samples of diferent grades. Here, we intersected genome‑wide atlas data with topologically associating domains (TADs) and demonstrated that the chromatin organization and epigenetic landscape of enhancers have a strong impact on genes diferentially expressed in WHO low grade versus high grade gliomas. We identifed TADs enriched in glioma grade‑specifc genes and/or epigenetic marks. We found the set of transcription factors, including REST, E2F1 and NFKB1, that are most likely to regulate gene expression in multiple TADs, containing specifc glioma‑related genes. Moreover, many genes associated with the cell–matrix adhesion Gene Ontology group, in particular 14 PROTOCADHERINs, were found to be regulated by long‑range contacts with enhancers. Presented results demonstrate the existence of epigenetic diferences associated with chromatin organization driving diferential gene expression in gliomas of diferent malignancy. Gliomas are primary brain tumors originating from neural stem cells or progenitor cells1. Tey range from benign and highly-curable pilocytic astrocytomas (World Health Organisation, WHO grade I, GI), through difuse astrocytomas that could be benign (WHO grade II, GII) or malignant (WHO grade III, GIII) to highly malig- nant glioblastomas (WHO grade IV, GIV)2. -

Downloaded from ENCODE and Aligned to the Human Genome (Grch38 Release 95) Using Hisat2 (V2.1.0)
UCSF UC San Francisco Electronic Theses and Dissertations Title miRNA-independent function of lnc-pri-miRNA loci Permalink https://escholarship.org/uc/item/6rc5w9fw Author He, Daniel Publication Date 2021 Peer reviewed|Thesis/dissertation eScholarship.org Powered by the California Digital Library University of California by Submitted in partial satisfaction of the requirements for degree of in in the GRADUATE DIVISION of the UNIVERSITY OF CALIFORNIA, SAN FRANCISCO Approved: ______________________________________________________________________________ Chair ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ ______________________________________________________________________________ Committee Members ii Acknowledgements I am first and foremost extremely lucky and grateful to have family that is supportive of my academic endeavors throughout my life. As the first of my family to graduate college, I’ve learned the true value of hard-work, determination, and love through my parents. They continue to serve as inspiration for me to grow and become a better version of myself every day. I am incredibly grateful to have had the opportunity to learn and grow as a scientist in the laboratory of Dr. Daniel Amos Lim. Dr. Lim has an unparalleled work-ethic that is motivating and inspiring for his students and mentees. I can truly say that I’ve never worked with a mentor that is so meticulously involved with the growth and success of his mentees. These past 5 years in Dr. Lim’s lab will remain with me forever, and I will never forget the lessons I’ve learned about how to be a good scientist, and a good person. Without a doubt, the academic environment within Dr. Lim’s lab is mentally stimulating – where research spans from fine-grain detail of molecular biology, to broader impact health-care tools. -

Elucidating the Constitutional Genetic Basis of Multiple Primary Tumours
Elucidating the constitutional genetic basis of multiple primary tumours James William Whitworth Gonville and Caius College February 2019 This dissertation is submitted for the degree of Doctor of Philosophy Preface This dissertation is the result of my own work and includes nothing which is the outcome of work done in collaboration except as declared in the Preface and specified in the text. It is not substantially the same as any that I have submitted, or, is being concurrently submitted for a degree or diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text. I further state that no substantial part of my dissertation has already been submitted, or, is being concurrently submitted for any such degree, diploma or other qualification at the University of Cambridge or any other University or similar institution except as declared in the Preface and specified in the text It does not exceed the prescribed word limit for the relevant Degree Committee. Acknowledgments I would like to extend my sincere thanks to the following people. To my supervisor, Eamonn Maher, for his mentorship over a time period longer than the programme of study outlined in this thesis. I am also grateful to Marc Tischkowitz, my second supervisor, for his encouragement and input. To my colleagues past and present in the Academic Department of Medical Genetics for their insights, collaboration, advice, and camaraderie over the past few years. In particular Ruth Casey, Graeme Clark, France Docquier, Ellie Fewings, Benoit Lang-Leung, Ezequiel Martin, Eguzkine Ochoa, Faye Rodger, Phil Smith and Hannah West. -

The Genome of the Vervet (Chlorocebus Aethiops Sabaeus)
Downloaded from genome.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press The genome of the vervet (Chlorocebus aethiops sabaeus) Wesley C. Warren1, Anna J. Jasinska2, 3, Raquel García-Pérez4, Hannes Svardal5, Chad Tomlinson1, Mariano Rocchi6, Nicoletta Archidiacono6, Oronzo Capozzi6, Patrick Minx1, Michael J. Montague1, Kim Kyung1, LaDeana W. Hillier1, Milinn Kremitzki1, Tina Graves1, Colby Chiang1, Jennifer Hughes7, Nam Tran2, Yu Huang2, Vasily Ramensky2, Oi-wa Choi2, Yoon J Jung2, Christopher A. Schmitt2, Nikoleta Juretic8, Jessica Wasserscheid8, Trudy R. Turner9,10, Roger W. Wiseman11, Jennifer J. Tuscher11, Julie A. Karl11, Jöern E. Schmitz12, Roland Zahn13, David H. O’Connor11, Eugene Redmond14, Alex Nisbett14, Béatrice Jacquelin15, Michaela C Müller-Trutwin15, Jason M Brenchley16, Michel Dione17, Martin Antonio17, Gary P. Schroth18, Jay R. Kaplan19, Matthew J. Jorgensen19, Gregg W.C. Thomas20, Matthew W. Hahn20, Brian J. Raney21, Bronwen Aken22, Juergen Schmitz23, Gennady Churakov23,24, Angela Noll23, Roscoe Stanyon25, David Webb26, Francoise Thibaud-Nissen26, Magnus Nordborg5, Tomas Marques- Bonet4, Ken Dewar8, George M. Weinstock27, Richard K.Wilson1, Nelson B. Freimer2 1The Genome Institute, Washington University School of Medicine, St Louis, MO 63108, USA. 2Center for Neurobehavioral Genetics, Semel Institute for Neuroscience and Human Behavior, University of California Los Angeles, 90095, CA, USA. 3Institute of Bioorganic Chemistry, Polish Academy of Sciences, Poznan, Poland. 4ICREA at Institut de Biologia Evolutiva, (UPF-CSIC) and Centro Nacional de Analisis Genomico (CNAG), PRBB/PCB, 08003 Barcelona, Spain. 5Gregor Mendel Institute, Austrian Academy of Sciences, Vienna Biocenter (VBC), 1030 Vienna, Austria. 6Department of Biology, University of Bari, Bari, Italy. Downloaded from genome.cshlp.org on September 27, 2021 - Published by Cold Spring Harbor Laboratory Press 7Whitehead Institute, Cambridge, Massachusetts 02142, USA. -

Identificação De Mutações Germinativas Em Pacientes Com Câncer Papilífero Familiar De Tireoide Através De Análise De Exoma
UNIVERSIDADE FEDERAL DE MINAS GERAIS PROGRAMA DE PÓS-GRADUAÇÃO EM MEDICINA MOLECULAR IDENTIFICAÇÃO DE MUTAÇÕES GERMINATIVAS EM PACIENTES COM CÂNCER PAPILÍFERO FAMILIAR DE TIREOIDE ATRAVÉS DE ANÁLISE DE EXOMA Débora Chaves Moraes Ribeiro Orientador: Prof. Luiz Armando Cunha de Marco Co-orientadora: Profª. Luciana Bastos Rodrigues Belo Horizonte 2018 IDENTIFICAÇÃO DE MUTAÇÕES GERMINATIVAS EM PACIENTES COM CÂNCER PAPILÍFERO FAMILIAR DE TIREOIDE ATRAVÉS DE ANÁLISE DE EXOMA Débora Chaves Moraes Ribeiro Débora Chaves Moraes Ribeiro IDENTIFICAÇÃO DE MUTAÇÕES GERMINATIVAS EM PACIENTES COM CÂNCER PAPILÍFERO FAMILIAR DE TIREOIDE ATRAVÉS DE ANÁLISE DE EXOMA Tese apresentada ao Programa de Pós-graduação em Medicina Molecular da Universidade Federal de Minas Gerais, como requisito para qualificação no doutorado em Medicina Molecular Área de concentração: Medicina Molecular Linha de pesquisa: Oncologia Orientador: Prof. Dr. Luiz Armando Cunha de Marco Co-Orientadora: Profª. Drª. Luciana Bastos Rodrigues Belo Horizonte Faculdade de Medicina da UFMG 2018 Dedico esse trabalho aos meus pais, ao Eugênio Ribeiro, ao João Guilherme, as minhas irmãs e ao Eduardo Galo. A eles, toda minha admiração, amor e gratidão. i AGRADECIMENTOS A Deus por todas as oportunidades de aprendizado e crescimento, e pela beleza e grandiosidade da vida; Aos meus pais, meus grandes exemplos de integridade, superação, virtude e compaixão, agradeço, com toda admiração e gratidão, pela convivência, força, amor, paciência, carinho e cuidado; Ao Eugênio, meu grande companheiro, pessoa